| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
GeNeViSTA
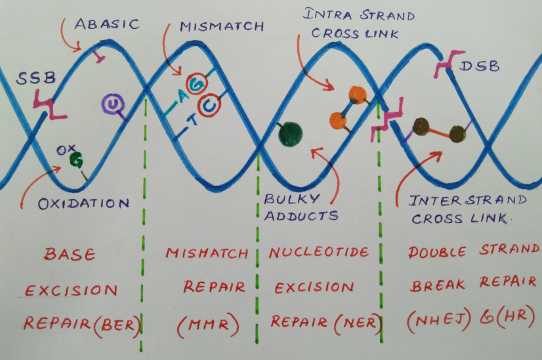
| DNA Repair Mechanism | Proteins/ protein complexes involved
|
| Base excision repair | DNA glycosylases, APEX1, XRCC1, PNKP, TDP1, APTX, DNA polymerases β, δ and ε, FEN1, PCNA, Replication factor subunits, Poly (ADP-Ribose) polymerases. |
| Mismatch repair | MutSα (MSH2-MSH6 heterodimer), MutSβ (MSH2-MSH3 heterodimer), MutLα (MLH1-PMS2 heterodimer), MutLβ (MLH1-PMS2 heterodimer), MutLγ (MLH1-MLH3 heterodimer), Exonuclease 1, PCNA, Replication factor subunits. |
| Nucleotide excision repair | XPC-Rad23B-CEN2 complex, DDB1-XPE complex, CSA, CSB, TFIIH, XPB, XPD, XPA, RPA, XPG, ERCC1- XPF, DNA polymerase δ or ε. |
| Double strand break repair: |
|
| - Homologous recombination | Mre11-Rad50-Nbs1 complex, CtIP, RPA, Rad51, Rad52, BRCA1, BRCA2, Exonuclease1, BLM-TopIIIα complex, GEN1-Yen1 complex, Slx1- Slx4 complex, Mus81/Eme1. |
| - Non-homologous end-joining | Ku70-Ku80 complex, DNA-PKc complex, XRCC4-DNA ligase IV complex, XLF. |
Germline mutations in the genes that code for the various proteins involved in the DNA repair pathways (listed in Table 1) result in different disorders, which are referred to as inherited disorders of DNA repair. The relatively common DNA repair disorders along with their molecular basis are listed in Table 2.
| Disorder | Genes | Molecular mechanism |
| | associated | |
| Disorders of Nucleotide Excision Repair
| ||
| Cockayne syndrome | ERCC8 ERCC6 | The proteins encoded by ERCC6 and ERCC8 help in transcription-coupled nucleotide excision repair (TC-NER) which removes ultraviolet radiation (UV)-induced pyrimidine dimers and other transcription-blocking lesions from the transcribed strands of the active genes. Mutations in these genes impair the TC-NER mechanism (Citterio et al., 2000). |
| Xeroderma pigmentosum | XPA XPB/ ERCC3 XPC XPD/ ERCC2 XPE/ DDB2 XPF/ERCC4 XPG/ ERCC5 XPV/ POLH
| The nucleotide excision repair genes XPB and XPD partially unwind the DNA in the region of the damage for further processing. The XPF product makes a single-strand nick at the 5’ side of the lesion and the XPG product makes a similar nick on the 3’ side, releasing a region of approximately 30 nucleotides containing the damage. The resulting gap is filled by DNA polymerase using the other (undamaged) strand as a template in a process involving proliferating cell nuclear antigen (PCNA). Mutations in these genes impair the global genome NER mechanisms (Arlett et al., 2006). |
| Trichothiodystrophy | ERCC2/ XPD ERCC3/ XPB GTF2H5 MPLKIP RNF113A GTF2E2
| XPB and XPD encode subunits of the TFIIH (transcription factor II H) complex, which is included in nucleotide excision repair, in RNA polymerase (RNA pol I & pol II) transcription initiation and regulation, and in cell cycle control. TFIIH opens the double-stranded DNA around the defect, through the helicase activity of XPD and the ATPase activity of XPB, the core subcomplex associates with NER-specific factors, including XPA, and mediates incision/excision of the damaged oligonucleotide. Mutations in these genes impair the global genome NER mechanisms. |
| Cerebro-oculo-facial-skeletal (COFS) syndrome | ERCC6 ERCC2 ERCC5 ERCC1
| Similar mechanism as described above for Cockayne syndrome and Xeroderma pigmentosum. |
| Disorders of DNA Mismatch Repair
| ||
| Hereditary nonpolyposis colorectal carcinoma (Lynch Syndrome)
Muir Torre Syndrome – subtype of Lynch Syndrome | MLH1 MSH2 MSH6 PMS2 EPCAM
| Germline pathogenic variant in a mismatch repair gene causes microsatellite instability (MSI) and disrupts the mismatch repair (MMR) pathway |
| Disorders of Double Strand Break Repair
| ||
| Ataxia telangiectasia | ATM | The ATM protein assists cells in recognizing double stand DNA breaks and activates enzymes which repair the breaks in the strand. Without this protein, cells become unstable and undergo apoptosis. |
| Nijmegen breakage syndrome | NBN | The NBN protein product nibrin directs the NBN/Mre11/Rad50 (MRN) complex to the sites of double-strand breaks and interacts with ATM kinase to coordinate cell cycle arrest with DNA repair. Mutation in the NBN gene impairs the formation of the MRN complex (Carney et al., 1998). |
| Others
| ||
| Bloom Syndrome
| RECQL3 |
The
RecQ
helicases
unwind
the
double
helix
of
the
DNA
molecule
during
replication
as
well
as
DNA
repair.
Without
these
helicases,
the
cell
is
not
able
to
efficiently
repair
the
DNA
damage
caused
by
ultraviolet
light
and
other
damaging
agents.
Mutations
in
these
genes
lead
to
an
increase
in
the
frequency
of
sister
chromatid
exchanges
(around
10
times
higher
than
average)
and
increase
in
the
frequency
of
chromosome
breakages
(Monat,
2010;
Friedrich
et
al.,
2010). |
| Rothmund-Thomson syndrome | RECQL4 |
|
| Werner syndrome
| RECQL2 |
|
| Fanconi Anemia | FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, BRCA2, BRIP1 | The proteins produced by these genes form the FA pathway which is involved in the repair of DNA damage caused by interstrand cross links (Dutrillaux et al., 1982). |
Most of the inherited disorders of DNA repair are associated with a strong predisposition to malignancies (Tomaszewska et al., 2006). Additionally, many of them have associated immunodeficiency, which could be attributable to the fact that DNA repair proteins are involved in joining the double-stranded breaks between the variable (V), diversity (D), and joining (J) segments of the lymphocyte immunoglobulin and antigen receptor genes. Features of ‘early aging’ (progeroid characteristics) are also seen in many of the disorders in this group. The clinical features of some of the common DNA repair defects are listed hereunder.
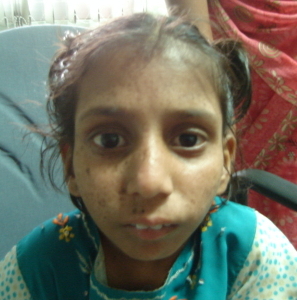
Cockayne syndrome (CS) is a spectrum of disorders. Patients with CS type I (classic or moderate type) have growth retardation, global developmental delay and microcephaly since infancy, and thereafter develop cachectic dwarfism, cutaneous photosensitivity, pigmentary retinopathy, cataracts, sensorineural hearing impairment, and progressive central and peripheral nervous dysfunction including demyelinating peripheral neuropathy and leukodystrophy (Figure 2a & 2b). CS type II (severe or early-onset CS) manifests as growth failure from the intrauterine period with severe postnatal neurologic impairment and profound global developmental delay. There is accelerated development of kyphoscoliosis and other joint abnormalities. CS type III (mild or late-onset CS) has normal growth and cognitive development with late onset symptoms. Management includes physical therapy, antispasticity medications, use of sun screens and sun glasses for skin and lens/retina sensitivity, dental care, and management of hearing loss and cataract.
Xeroderma pigmentosum (XP) presents with freckles over of the face during infancy, excessive sun sensitivity (sunburn with blistering, persistent erythema) with marked, sunlight-induced ocular involvement (photophobia, keratitis) and high risk of sunlight-induced cutaneous neoplasms like basal cell carcinoma, squamous cell carcinoma and melanoma (Greenhaw et al., 1992). Around one-fourths of the affected individuals have acquired microcephaly, progressive hearing loss, and cognitive impairment. Death is commonly due to skin cancer and neurologic degeneration. XP associated with severe neurologic impairment, stunted growth and delayed secondary sexual development is called the DeSanctis-Cacchione variant.
Classic ataxia-telangiectasia (A-T) also known as Louis Bar syndrome manifests as progressive cerebellar ataxia, oculomotor apraxia, choreoathetosis, telangiectasias of the conjunctivae, and immunodeficiency causing frequent infections (Lin et al., 2014). There is increased risk of hematological malignancies. The initial symptom is usually cerebellar ataxia which starts around 1 to 4 years of age and majority of them become wheelchair bound by adolescence. A-T patients are highly sensitive to ionizing radiation, like medical X rays. The non-classic forms of A-T include adult-onset A-T and A-T with early-onset dystonia. Apart from physical therapy and supportive care, monitoring for severe infections and malignancies and aggressive treatment of infections, with IVIG wherever required, is recommended.
People with this condition may have bone marrow failure, physical abnormalities like radial ray defects, short stature, organ defects like malformed or absent kidneys, other defects of the urinary tract, gastrointestinal abnormalities, cardiac defects and an increased risk of certain malignancies. Skin may show hyperpigmentation or café-au-lait spots. Other symptoms are small abnormally shaped eyes and malformed ears with hearing loss. They may have abnormal internal and external genitalia and could be infertile. Central nervous system abnormalities include hydrocephalus and microcephaly. There is excessive risk of developing malignancies like acute myeloid leukemia and tumors of skin, gastrointestinal system, genital tract and tumors of head and neck. Patients with symptomatic anemia may require blood transfusions. Erythropoetin and colony stimulating factors could increase the blood count and give temporary relief, but bone marrow transplantation is usually required to definitively manage the aplastic anemia. Caution is required in the management of these patients as they have an increased risk of adverse events and toxicity related to chemotherapeutic and immunosuppressive therapies. Though most types of Fanconi anemia have an autosomal recessive inheritance pattern, RAD51-related FA has an autosomal dominant inheritance pattern and FANCB-related FA is inherited in an X-linked manner.
Bloom Syndrome manifests as severe intrauterine and postnatal growth deficiency, loss of subcutaneous fat, short stature and sun-sensitive skin lesions of the face (Figure 3). Gastroesophageal reflux is common and causes recurrent respiratory tract infections. Learning disability is often seen. Affected males are usually infertile, while females may be fertile but usually have premature menopause. Chronic obstructive pulmonary disease, diabetes mellitus and increased risk of malignancies are also associated with this condition. The management is usually symptomatic and supportive.
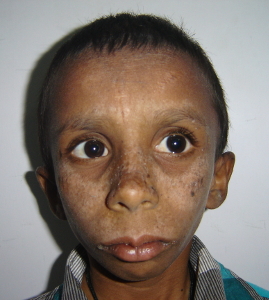
Nijmegen breakage syndrome manifests with growth retardation, short stature, progressive reduction in head circumference and developmental delay. There is associated progressive cognitive decline. Humoral immunodeficiency results in recurrent respiratory tract infections (Chrzanowska et al., 2012). There is increased risk of malignancies like lymphomas and also predisposition to tumors such as medulloblastoma and glioma. The immunodeficiency and malignancies often result in early deaths. Affected females may develop premature ovarian failure. Treatment is supportive and symptomatic. IVIG can be given for severe humoral immunodeficiency causing frequent infections. Vitamin E and folic acid supplementation are recommended. For malignancies, standard anti malignancy agents and hematopoietic stem cell transplantation are preferred. Periodic follow-up to monitor growth and development and management of frequent infections is required for these patients.
Patients with Lynch syndrome develop colorectal cancer and other HNPCC-typical tumors like stomach, small intestine, endometrial carcinomas, cervical, breast cancers, bladder cancers (Lynch & Lynch, 2005). Periodic surveillance of people with HNPCC with colonoscopy and surgical removal of the tumor is the available management at present. The Muir-Torre variant of Lynch syndrome presents with neoplasms of the skin like sebaceous adenomas, sebaceous carcinomas, and keratoacanthomas along with the other internal malignancies usually associated with Lynch syndrome. Unlike the other DNA repair disorders, Lynch syndrome has an autosomal dominant pattern of inheritance and presymptomatic screening of at-risk family members is essential, for appropriate surveillance and presymtomatic therapeutic intervention of the other mutation – carrying family members (Hegde et al., 2014).
Trichothiodystrophy (TTD) is characterized by brittle, sulfur-deficient hair that shows a typical trichorrhexis nodosa pattern on light microscopy and an alternating light and dark banding pattern called ‘tiger tail banding’ under polarizing microscopy (Figure 4). Patients with TTD can have a wide variety of clinical features including cutaneous manifestations, neurologic impairment, and growth abnormalities. Ichthyosis, intellectual disability, reduced fertility, ocular abnormalities, short stature, and recurrent infections are associated features. The disorder has both photosensitive and non-photosensitive forms. Unlike the other DNA repair disorders, patients with TTD have not been reported to have a predisposition to cancer.
This condition is associated with poikiloderma congenita, growth retardation, alopecia, photosensitivity, nail dystrophy, teeth anomalies, cataract and hypogonadism. Skeletal anomalies such as radial ray defects, ulnar defects, absent or hypoplastic patella, and/ or osteopenia may occur in these patients (Figure 5). They have an increased risk of skin cancers and other malignancies. Management includes pulsed dye laser to treat the telangiectatic component of the rash, surgical removal of cataracts and treatment for malignancies as per standard protocols. Use of sunscreens with both UVA and UVB protection is recommended to prevent skin cancers and calcium and vitamin D supplements are recommended for osteopenia.
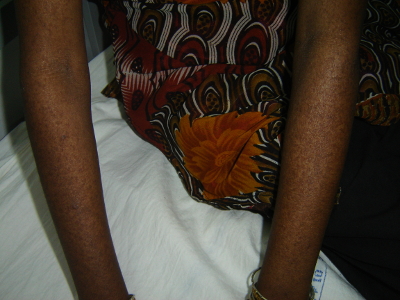
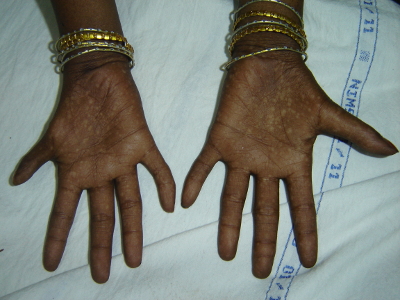
Werner syndrome is characterized by signs of premature ageing and predisposition to cancer. Symptoms usually start after the second decade of life with graying of hair, excessive hair loss, voice change, short stature and scleroderma-like skin lesions. Later, the patients develop cataract, hypogonadism, diabetes, ulcers of skin, atherosclerosis and osteoporosis by around 30 years. Myocardial infarction and malignancies are the common causes of death. The mainstay of management is control of diabetes mellitus, use of cholesterol-lowering drugs for abnormal lipid profile, treatment of skin ulcers, surgical treatment of cataracts and management of malignancies as per standard protocols.
The diagnosis of these condition is established by a combination of clinical examination findings, laboratory investigations, and definitive molecular genetic testing. Dysmorphology evaluation and detailed systemic examination may offer important clues to the diagnosis as mentioned in the individual disease sections above.
Chromosomal breakage studies have conventionally been used for the diagnosis of disorders such as Fanconi anemia and Nijmegen breakage syndrome, which are associated with an increased number of cross linking agent-induced chromosomal breakages. For this study, T lymphocytes in a peripheral blood sample are cultured in the presence of a cross-linking agent such as Mitomycin-C or Diepoxybutane at varying concentrations, after which chromosomal aberrations are quantified in metaphase spreads. The chromosomal aberrations seen include chromatid gaps, chromatid breaks, tri and quadri radials, and complex interchange figures (Figure 6). More than ten breaks per cell is usually considered significant and is strongly suggestive of a chromosomal breakage syndrome (Anneke et al., 2012).

Courtesy: Dr Shubha R Phadke, Professor & Head, Department of Medical Genetics, SGPGIMS, Lucknow.
Flow cytometry is now a preferred alternative test for studying chromosomal breakages, since it does not require setting up of cultures and has a rapid turn-around time. It also eliminates the need for technical expertise and rules out intra-observer variations seen in scoring. It is based on the principle that cells are arrested at late S/early G2 phase of the cell cycle when exposed to cross linking agents and therefore the fraction of arrested cells can be taken as a measure of their sensitivity to cross linking agents.
Definitive diagnosis is most often possible through molecular genetic testing of the known associated genes. As most of these disorders are genetically heterogeneous and the causative genes are large genes, next generation sequencing-based multigene panel testing is the preferred modality for molecular diagnosis, unless the patient is from an ethnic population, with a high frequency carrier rate for a specific mutation, where a targeted testing may be applied. Presymptomatic testing and prenatal diagnosis may be offered to at-risk families, as applicable, once the underlying disease-causing mutations are identified in the proband.
There is no definite disease-specific curative therapy available for these disorders at present. Symptomatic and supportive treatment remains the mainstay of management of these disorders. Aggressive treatment is essential for the recurrent infections resulting from immunodeficiency and intravenous immunoglobulin (IVIG) may be required in severe infections. Surveillance for and early detection and early surgical/ medical management for malignancies is required for most of these conditions. Conditions like Fanconi anemia and Ataxia telangiectasia are associated with higher toxicity and adverse reactions to chemotherapy and radiotherapy and HSCT-related immunomodulatory regimens; therefore, caution and appropriate dose-adjustment is required for the same. Conditions with photosensitivity and sunlight-damage such as Xeroderma pigmentosum, Cockayne syndrome and Rothmund-Thomson syndrome require use of sunscreens and protective clothing, and avoidance of sun exposure.
Gene therapy trials are ongoing for Fanconi anemia and a Phase I study of the antioxidant quercetin in children with Fanconi anemia is also currently underway. In one study on patients with Xeroderma pigmentosum, the bacterial DNA repair enzyme T4 endonuclease V in a topical liposome-containing preparation has been found to reduce the frequency of new actinic keratoses and basal cell carcinomas (Yarosh et al., 2001), but this treatment has not yet been approved by the US Food and Drug Administration (FDA). Oral vismodegib, an inhibitor of the hedgehog pathway, has been approved by the FDA for treatment of metastatic basal cell carcinoma or locally advanced basal cell carcinoma that has recurred following surgery or in individuals not fit for surgery or for radiation therapy. For ataxia telangiectasia, antioxidants (vitamin E or alpha-lipoic acid) are recommended, although no formal testing for efficacy has been conducted in affected individuals. Stop codon read-through with aminoglycosides and Antisense morpholino oligonucleotides have shown some therapeutic benefit in animal models of A-T. Steroid therapy with dexamethasone or betamethasone, especially when delivered by loading into erythrocytes, has been found to be beneficial in reducing the neurological manifestations of A-T and Phase III clinical trials are ongoing for this modality (Leuzzi et al., 2015).
As most of the conditions in this group have an autosomal recessive pattern of inheritance, the parents will usually be obligate carriers and the recurrence risk in siblings of affected individuals would be 25%. Lynch syndrome has an autosomal dominant pattern of inheritance with a 50% risk of recurrence in each offspring and sibling of an affected individual and therefore appropriate pre-symptomatic testing and surveillance of at-risk family members is essential. Female heterozygous carriers of ataxia telangiectasia-related ATM gene mutation and the Fanconi anemia-related FANCD2, FANCN and FANCJ gene mutation have increased susceptibility to breast cancer and should be recommended surveillance for the same. In addition, heterozygous carriers of ATM gene mutation can have increased radiation sensitivity and increased risk of toxicity of radiotherapy and higher-dose diagnostic radiation exposure and should be warned about the same.
DNA repair disorders are a genetically heterogeneous group of diseases and accurate diagnosis of these conditions can help in appropriate surveillance and management of not just the affected individuals but also their family members.
1. Anneke B, et al. Diagnosis of Fanconi Anemia: Chromosomal Breakage Analysis. Anemia 2012: 238731. doi: 10.1155/2012/238731.
2. Arlett CF, et al. Clinical and cellular ionizing radiation sensitivity in a patient with xeroderma pigmentosum. Br J Radiol 2006; 79: 510-7.
3. Carney JP, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 1998; 93: 477-86.
4. Chrzanowska KH, et al. Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis 2012; 7: 13.
5. Citterio E, et al. ATP-Dependent Chromatin Remodeling by the Cockayne Syndrome B DNA Repair-Transcription-Coupling Factor. Mol Cell Biol 2000; 20: 7643-53.
6. Clancy S. DNA damage & repair: mechanisms for maintaining DNA integrity. Nature Education 2008; 1:103.
7. Dexheimer TS. DNA repair pathways and mechanisms. In: DNA Repair of Cancer Stem Cells Mathews LA et al. (eds.). doi: 10.1007/978-94-007-4590-2_2, © Springer Science + Business Media Dordrecht 2013.
8. Dutrillaux B, et al. The cell cycle of lymphocytes in Fanconi anemia. Human Genet 1982; 62: 327-32.
9. Friedrich K, et al. WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum Genet 2010; 128:103-11.
10. Greenhaw GA, et al. Xeroderma pigmentosum and Cockayne syndrome: overlapping clinical and biochemical phenotypes. Am J Hum Genet1992; 50: 677-89.
11. Hegde M, et al. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Genet Med 2014;16: 101-16.
12. Leuzzi V, et al. Positive effect of erythrocyte-delivered dexamethasone in ataxia-telangiectasia. Neurol Neuroimmunol Neuroinflamm 2015; 2: e98.
13. Lin DD, et al. Cerebral Abnormalities in Adults with Ataxia-Telangiectasia. AJNR Am J Neuroradiol 2014; 35: 119-23.
14. Lynch HT, Lynch JF. What the physician needs to know about Lynch syndrome: an update. Oncology (Williston Park) 2005; 19: 455-63.
15. Monnat RJ Jr. Human RECQ helicases: Roles in DNA metabolism, mutagenesis and cancer biology. Semin Cancer Biol 2010; 20: 329-39.
16. Tomaszewska A, et al. Chromosome instability syndromes. Pol Merkur Lekarski 2006; 20: 577-81.
17. Yarosh D, et al. Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xeroderma Pigmentosum Study Group. Lancet 2001; 357: 926-9.
| Abstract | Download PDF |
 a)
a) 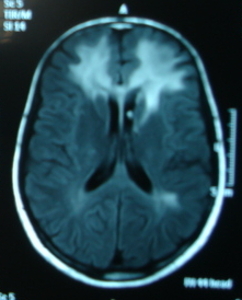 b)
b)
