Compound heterozygosity for p.F508del mutation and deletion of exons 4-11 in CFTR in an infant with cystic
fibrosis- Limitations of ARMS-PCR and Sanger sequencing
1Department of Clinical Genetics, Christian Medical College, Vellore, Tamil Nadu 2Department of Child Health, Christian Medical College, Vellore, Tamil Nadu Email:sdanda@cmcvellore.ac.in
Summary
Cystic fibrosis (CF) is a life threatening genetic condition caused due to mutations in the CFTR gene. This is an
autosomal recessive condition and usually parents are carriers of a heterozygous mutation in the CFTR gene. We describe
an infant diagnosed with the severe phenotype of CF and detected to be homozygous for p.F508del by ARMS-PCR.
However, parental studies confirmed carrier state in mother but the father showed a wild allele by ARMS-PCR
and sequencing. This prompted us to look for uniparental disomy (UPD) and large deletions. UPD was
ruled out by studying both intragenic and extragenic markers for chromosome 7. The proband and the
father were found to have a large deletion detected by MLPA involving exons 4-11 in the CFTR gene which
includes the region coding for phenylalanine at position 508. This case highlights the need of caution when
interpreting results of molecular genetic testing during genetic counseling. It is the first documented case from
India with point mutation and large deletion of the CFTR gene giving rise to apparent homozygosity for
p.F508del.
Case Report
A 5 month old boy diagnosed with CF was referred to the Clinical Genetics Unit of Christian Medical College, Vellore by
the Department of Child Health for molecular confirmation and genetic counselling. He was the first offspring of
non-consanguineous parents born through LSCS. He weighed 2.5 kgs at birth and was admitted in the
nursery for one week with respiratory distress probably secondary to meconium aspiration and managed
conservatively.
Previous history of recurrent hospital admissions for pneumonia, passage of oily loose stools and failure to thrive was
obtained from the parents. The current admission was for severe pneumonia leading to unstable vital signs and probable
hypoxic seizures requiring ventilation.
He had achieved partial head control and had been immunised till the age of 6 weeks. The family history was
unremarkable.
His length at 55cms and weight at 2.5kgs, were <3SDs below the age related mean; the head circumference at 38 cms
was normal. There were no dysmorphic features. Chest examination revealed crepitations and conducted sounds with
decreased bilateral air entry. The liver was palpable 1 cm below the right costal margin and rest of the
physical examination was normal. The child was treated with infusion of antibiotics and antiepileptics. Stool
examination showed numerous fat globules. He was started on nasogastric feeds on day 4 of hospitalization.
His high grade fever persisted and he deteriorated despite all supportive measures and succumbed to his
illness.
Molecular analysis was done for confirmation of diagnosis and genetic counseling. Preliminary genetic
counseling was given and parents agreed for carrier testing as they were considering prenatal diagnosis for future
pregnancies.
a)
b)
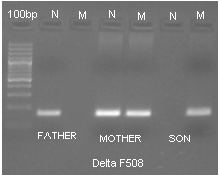
Figure 1: a). Gel picture of ARMS-PCR of proband and parents showing normal father, heterozygosity of del
F508 for mother and homozygosity of del F508 for proband. N- normal M- Mutant.
b). Sequencing chromatogram showing homozygosity for p.F508del in proband, heterozygosity in mother but
normal result in father.
Material and Methods
Genomic DNA was extracted from leukocytes using QIAGEN Midi Kit. ARMS-PCR was done for the p.F508del
mutation.1 Sequencing was done (Applied Biosystems Genetic Analyzer 3500) to confirm the mutations detected by
ARMS-PCR. Further molecular analysis was done using four intragenic (T854T, M470V, TUB9, TUB 20) and two
extragenic (XV 2C, KM19) markers for chromosome 7. Specific primers were used according to previously described
methods and the amplified products were restriction digested using TaqI, PstI, MnlI, HphI, AvaII, PvuII for XV2C,
KM19, TUB9, M470V, T854T and TUB20 respectively.2 The products of XV2C, KM19, M470V, T854T were separated
using 2% agarose gel and TUB 9 and TUB 20 were separated using 8% native polyacrylamide gel electrophoresis (PAGE).
Six VNTR markers were studied to rule out false paternity. To identify larger deletions affecting whole exons of the CFTR
gene, the sample was sent to the Institute of Human Genetics, University Hospital, Technical University
RWTH Aachen, Aachen, Germany where commercially available MLPA (multiplex ligation dependent probe
amplification) assay for CF (P091; MRC Holland/Amsterdam/NL) was used according to the manufacturer’s
instructions.
Results
a)
b)
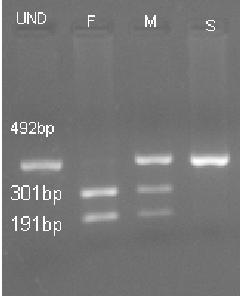
Figure 2: a). Markers status for M470V (HphI digest) F- Father’s alleles 2/2 M- Mother’s alleles 1/2 S- Son’s
alleles 1/1 UND– Undigested Product.
b). TUB 9 marker study (MnlI digest) by PAGE Well 1–100bp ladder. The digested product of TUB 9 (g.1525-61
A or G) using MnlI yields four fragments 199bp, 147bp, 130bp and 15bp for allele 1 and five fragments at 147bp,
130bp, 104bp, 95bp and 15bp for allele 2. F- Father has allele 2 which has 5 fragments - 147, 130, 104 bp are seen
and the smaller fragments (95 and 15 bp) which are not seen. M- Mother has 1 & 2 alleles (total 6 fragments)
-199, 147, 130, 104 bp are seen and the two smaller fragments are not seen. S- Son has allele 1/1 showing 199,
147, 130 bp; the 104 bp is not seen. H- Healthy.
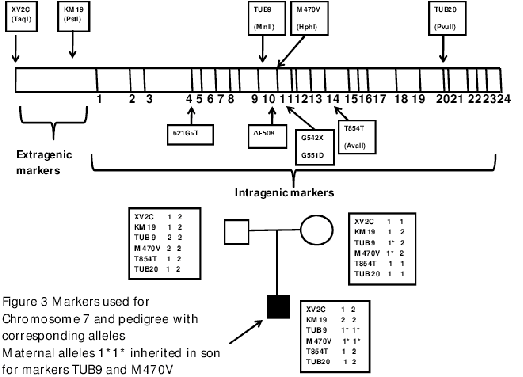
Figure 3: Markers used for Chromosome 7 and pedigree with corresponding alleles. Maternal alleles 1*1*
inherited in son for markers TUB9 and M470V.
The mutation analysis for CFTR by ARMS-PCR3 showed a pattern consistent with homozygous mutation
for p.F508del. Genetic testing of the parents showed only mother as carrier and father had the wild type
allele (Fig 1a). Sequencing results confirmed the pattern seen by ARMS-PCR (Fig 1b). Six VNTR markers
confirmed the paternity. Intragenic and extragenic markers for chromosome 7 showed presence of both
parental alleles for XV2C, KM19 T854T and TUB20. However the results for markers TUB9 and M470V
were not compatible with paternal inheritance as only maternal alleles were detected. (Figure 2a,2b) The
interpretation is as follows: the digested product of M470V (g.1540 A>G) using HphI yields one fragments at
492bp for homozygous allele 1, three fragments at 492bp, 301bp and 191bp for heterozygous allele 1 and
allele 2, and two fragments at 301bp and 191bp for (homozygous allele 2). The digested product of TUB 9
(g.1525-61 A or G) using MnlI yields four fragments 199bp,147bp, 130bp and 15bp for homozygous allele 1,
four fragments at 199bp, 147bp,130bp and 95bp (for heterozygous allele 1 and 2) and five fragments at
147bp,130bp,104bp,95bp and 15bp in the case of homozygous allele 2. Thus UPD was ruled out but deletion/
chromosomal rearrangement was suspected due to presence of only maternal alleles located near exon 10 of
CFTR. DNA analysis by MLPA at the Institute of Human Genetics in Aachen revealed that the proband
carried a large scale heterozygous deletion spanning exons 4-11. The same deletion was detected in the
father.
Discussion
Over 1900 different mutations have been identified in the CFTR gene (www.genet.sickkids.on.ca/). The most common
mutations are c.1521_1523delCTT (p.F508del), c.1652G>A, c.1641G>T and c.489+1G>T.3 The mutation p.F508del
(c.1521_1523delCTT), a deletion of three nucleotides resulting in removal of phenylalanine at position508, is the cause of
CF in majority of cases. It has a frequency of 66% in cases of CF worldwide.4 One study showed that affected individuals
of Indian origin have a frequency of p.F508del mutation ranging from 19% to 44%., while another Indian study showed 20
CF cases out of 100 to be homozygous for p.F508del and 13 cases to be heterozygous for this mutation.5,6 Cases with the
p.F508del mutation had a more severe phenotype. Single or multi exonic large deletion may be present in combination
with point mutations in CFTR on rare occasions.7 These can account for 1% to 3% of affected individuals.8 In the
present case either maternal UPD or a large gene rearrangement was suspected when routine molecular
methods showed apparent homozygosity for p.F508del, which was incompatible with the parental genetic
status.
A recent study showed that the frequency of large scale gene deletions differs from population to population. The
actual frequency of large scale deletion of the CFTR gene is still not clear since routine diagnostic analysis involves
detection of point mutations and small indels only and large scale deletions can be missed out. Advanced techniques like
MLPA, quantitative PCR and array CGH (comparative genomic hybridization) can be used to rule out the
apparent homozygosity of point mutations and indels.9 Chevalier-Porst et al. reported 24% large deletions in a
cohort of 1600 CF alleles tested. This French study reported two patients, a boy diagnosed with classical
CF at one year harbouring a deletion of 95.738 kb size which removed the coding sequence from exon 2
to exon 10, and a girl with reported deletion of exon 4–10. Both the cases had the p.F508 mutation in
the second allele inherited from the mother while the large deletion was inherited from the father, as in
the present case.10 In this case two extragenic markers and four intragenic markers ruled out UPD. Two
intragenic markers near exon 10 showed both alleles to be maternal further increasing the suspicion of
deletion. MLPA confirmed a large scale heterozygous deletion involving exons 4–11. This genotype detected
was associated with a very severe phenotype in the proband leading to death in infancy. This is the first
such case from India described in literature proving the importance of rarer CFTR rearrangements which
can have implications for genetic counseling and prenatal diagnosis. This also highlights the limitations of
ARMS-PCR and Sanger sequencing in detecting large genomic deletions in many single gene disorders.
Hence, interpretation of molecular genetic results has to be carried out with caution while counseling a
family.
Acknowledgements
We thank Professor Thomas Eggermann, Institute of Human Genetics, University Hospital, Technical University RWTH
Aachen, Aachen, Germany for doing the MLPA studies. Fluid grant from CMC Vellore was used for part of the molecular
work.
References
1. Ferrie RM, et al. Am J Hum Genet 1992; 51: 251-62.
2. Dörk T, et al. Hum Genet 1992; 88: 417-25.
3. Scobie G, et al. Mol Hum Reprod 199; 3: 203-7.
4. Zielenski, J & Tsui, L. Ann Rev Genet 1995; 29: 777–807.
5. Sharma N, et al. Ann Hum Genet 2009; 73: 26-33.
 a)
a) 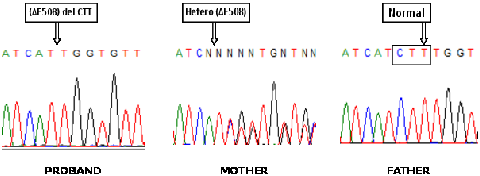 b)
b)
 a)
a) 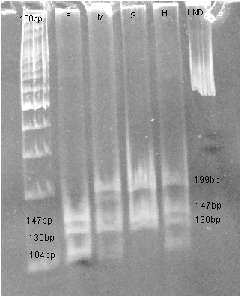 b)
b)