Superoxide Dismutase Deficiency due to Biallelic SOD1 Variants
Pallavi Sinha1, Sunita Bijarnia-Mahay1, Hemlata Wadhwani Bhatia2, Saurabh Chopra3 1Institute of Medical Genetics and Genomics, Sir Ganga Ram Hospital, New Delhi, India 2Department of Medical Genomics, Max Superspeciality Hospital, Shalimar Bagh, New Delhi, India 3Department of Pediatric Neurology, Apollo Hospital, Sarita Vihar, New Delhi, India Correspondence to: Dr Sunita Bijarnia-MahayEmail:Bijarnia@gmail.com
1 Abstract
Gain-of-function mutations in superoxide dismutase 1 (SOD1) are typically associated with familial Amyotrophic lateral
sclerosis (ALS). Recently a distinct neurodegenerative disorder has been described, occurring due to biallelic loss of
function in SOD1, manifesting as spastic tetraplegia with axial hypotonia in childhood. Debate exists regarding its
classification, as to whether it is a distinct disorder or a part of the ALS spectrum.
Superoxide dismutase (SOD) facilitates the transformation of the superoxide anion into hydrogen peroxide and oxygen
and plays an important role in the cellular antioxidant defense. In humans there are three different isoforms
of SODs: human Cu–Zn SOD (SOD1), the mitochondrial MnSOD (SOD2) and the extracellular Cu–Zn
SOD (SOD3). Impairment of their antioxidant function or overactivity due to gain-of-function molecular
mechanisms, represents a major pathophysiological role in the development of human neurodegenerative disorders
(primarily, Amyotrophic Lateral Sclerosis) and cancer linked to SOD1 abnormalities (Eleutherio et al.,
2021).
Mutations in SOD1 are known to be associated with familial autosomal dominant Amyotrophic Lateral Sclerosis
(ALS) mainly due to gain-of-function mechanism. Recently, Andersen et al. (2019), Park et al. (2019) and de Souza et
al. (2021) reported paediatric patients with a severe neuromuscular disorder characterized by progressive
motor neuron disease with hypotonia, spastic tetraplegia and loss of motor function due to homozygous
biallelic pathogenic variants in SOD1. With no family history of ALS conforming to autosomal dominant
inheritance, it was concluded that this disorder is distinct from SOD1- related ALS and constitutes a new clinical
entity.
We present the clinical and genetic findings of a patient with biallelic pathogenic variant in SOD1 associate with an
early neurodegenerative phenotype.
3 Patient Details
A 1-year-old male child, third born to consanguineous parents from Uzbekistan, was referred for genetic evaluation. He
was born at term gestation, by lower segment Cesarean section (LSCS), with a birth weight of 3.7 kg. The neonatal period
was uneventful. He achieved all developmental milestones at an appropriate age until the age of 7 months after which he
was noted to develop a gradual motor decline, with tightness of lower limbs being noted at around nine months of age. At
12 months of age, there was further motor regression with loss of ability to crawl or sit with support. There were no
concerns regarding social, communication and emotional responses. There was no history of seizures, and visual or hearing
impairment.
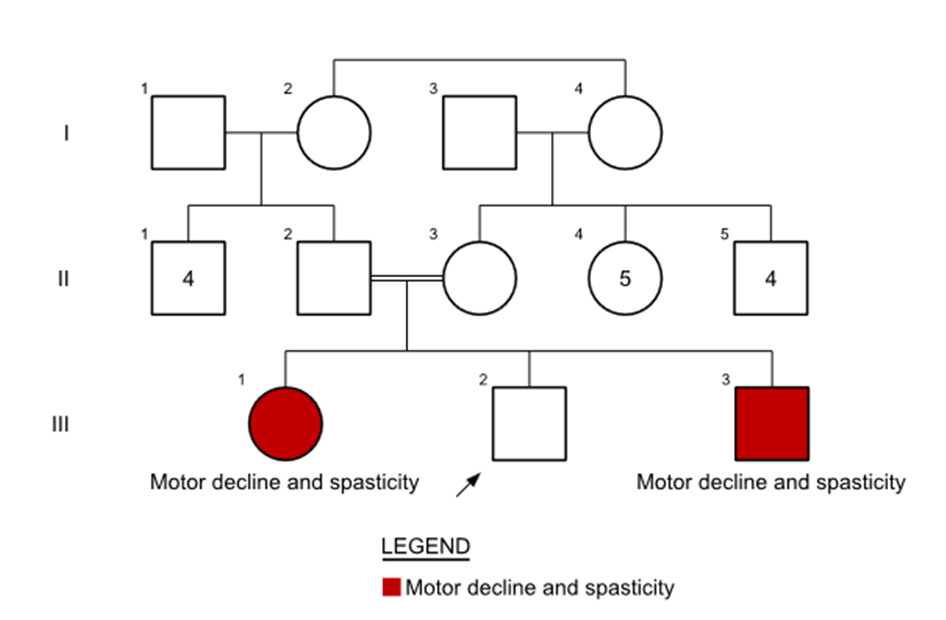
The proband’s elder sister, who is 10 years old, was reported to be similarly affected (Figure 1). Parents noted a
gradual motor decline for her, after 5-6 months of age. Currently, she was bedridden and was not brought for evaluation.
There was absence of speech in her. Social and communication domains were seemingly preserved. Magnetic resonance
imaging (MRI) of her brain showed evidence of cerebellar atrophy.
Figure 1: Figure 1: Pedigree of the family showing consanguinity and similar affected status of the elder sibling.
The parents were first cousins and were healthy and asymptomatic. There was no history of similar occurrence in the
extended family. Paternal grandparents, who had passed away, had no neurological symptoms. The maternal grandparents
were symptom free at the age of 70 years.
On examination, his growth parameters were within normal limits; weight: 10 kg (+0.33 Z), length: 75 cm (-0.32 Z),
and head circumference: 46cm (-0.05 Z). The general physical examination was unremarkable. On neurological
examination, the child was noted to be irritable and had appendicular hypertonia with axial hypotonia. Deep tendon
reflexes were brisk in all four limbs. Bilateral plantar response was extensor. No fasciculations, myokymia or hyperekplexia
were observed. Eye examination including fundus was unremarkable.
MRI brain was normal with no evidence of cerebral or cerebellar atrophy. In view of the progressive neurological
symptoms in the child as well as his elder sibling with history of consanguinity, an autosomal recessive genetic condition
was suspected and investigated for. Initial work up including hematological parameters, liver function tests,
kidney function tests and serum creatine phosphokinase (CPK) were normal. Metabolic work up, including
plasma lactate, homocysteine, acyl-carnitine, and amino acid profile by tandem mass spectrometry were also
normal.
Whole exome sequencing revealed a homozygous pathogenic variant in the SOD1 gene which is consistent
with a diagnosis of autosomal recessive spastic tetraplegia and axial hypotonia, progressive type (Table1).
This variant creates a shift in the reading frame starting at codon 112. The new reading frame introduces a premature
stop codon 10 positions downstream. This variant has previously been reported as disease-causing (Andersen et al.,
2019).
4 Discussion
The link between SOD1 variants and familial ALS is well-established, attributed to a gain of function in the SOD1 gene.
This leads to heightened oxidative activity, causing an overproduction of hydrogen peroxide. The mutated SOD1 also
promotes increased protein–protein interaction, fostering aggregation, dimer destabilization, and oligomerization. These
alterations contribute to abnormal axonal transport, microglia activation, heightened apoptosis, mitochondrial
dysfunction, and oxidative stress, ultimately playing a critical role in motor dysfunction (de Souza et al., 2021; Kaur et
al., 2016).
Biallelic truncating variants in SOD1 result in spastic tetraplegia with axial hypotonia. There are very few cases
reported in literature till now with the first case being reported in 2019 (Andersen et al., 2019). It is hypothesized that
complete loss of function of SOD1 enzyme activity can produce an increased vulnerability to oxidative stress with
mitochondrial dysfunction. It is characterized by onset of severe and progressive motor dysfunction in the first year of life.
There is severe axial hypotonia combined with spastic tetraplegia, hyperekplexia, hypertonia, extensor plantar response
and myokymia, reflecting upper motor neuron involvement. Cognitive development may be affected with absence of
speech. Andersen et al. (2019) described the autosomal recessive SOD1 gene-related disorder as a distinct entity, based on
clinical differences (early onset spastic tetraplegia with axial hypotonia) and loss-of-function mechanism of gene
malfunction. This was however challenged by de Souza et al. (2021), when they reported five cases from two
consanguineous families from Brazil, proposing it to be a varied spectrum of the same disorder. The patients reported by
de Souza et al. who had the same variant as our patient, also exhibited features of lower motor neuron
disease like fasciculations and fibrillations. They proposed that these patients may represent a very early
infantile-onset ALS. However, there is insufficient evidence to suggest the same and long-term studies are
required.
Regarding parental carriers who are heterozygous for the variant c.335dupG (p.Cys112Trpfs*11) in the SOD1 gene, it
is not possible to comment about the future risk of them developing a neurodegenerative disease, but they are not
expected to develop any symptoms of ALS due to the very nature of the variant (loss of function). However, a long-term
clinical follow-up would be necessary to detect any neurological manifestations related to ALS or any other type of
neurodegeneration.
The final word is yet awaited regarding the molecular mechanism of the disease as the pathophysiology continues to
unfold. However, it is clear that monoallelic as well as biallelic variants in the SOD1 gene should be studied carefully,
keeping in mind the clinical features as well as the mechanism of gene dysfunction created by the variant before making a
confirmed diagnosis.
1. Andersen PM, et al. Phenotype in an Infant with SOD1 Homozygous Truncating Mutation. N Engl J
Med. 2019; 381(5): 486-488.
2. de Souza PVS, et al. Progressive spastic tetraplegia and axial hypotonia (STAHP) due to SOD1 deficiency:
is it really a new entity? Orphanet J Rare Dis. 2021;16(1): 360.
3. Eleutherio ECA, et al. SOD1, more than just an antioxidant. Arch Biochem Biophys. 2021; 697: 108701.
4. Kaur SJ, et al. Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene. 2016;
577(2):109–118.
5. Park JH, et al. SOD1 deficiency: a novel syndrome distinct from amyotrophic lateral sclerosis. Brain. 2019;
142(8): 2230-2237.