Approach to a Child with Dysmorphism/ Congenital Malformation
Prajnya Ranganath
Department of Medical Genetics, Nizam’s Institute of Medical Sciences, Hyderabad Email:prajnyaranganath@gmail.com
Definition
Dysmorphology is a discipline of clinical genetics which deals with the study of abnormal patterns of human
growth and with the recognition and study of congenital human structural anomalies and patterns of birth
defects.
Congenital malformations/ birth defects can be sub-classified as major or minor anomalies.
Major anomalies are those that interfere with the normal functioning of an individual and pose a significant
health problem or risk to life. E.g. congenital heart defects, neural tube defects, omphalocele, cleft palate
etc.
Minor anomalies do not interfere with the normal functioning of an individual and usually are only of
cosmetic significance. E.g. simian crease, accessory nipple, clinodactyly, pre-auricular skin tag etc.
Major anomalies are present in 2-3% and minor anomalies are present in around 15% of live births. Minor anomalies are
usually associated with an increased risk of associated major anomalies and therefore presence of minor anomalies should
prompt a thorough search for associated major anomalies.
Classification of congenital anomalies
Congenital anomalies are classified, on the basis of the developmental stage in which the insult occurred, the process that
caused the change and the end result, into:
Malformation: Primary intrinsic developmental defect usually caused by genetic/ environmental/
multi-factorial causes (recurrence risk varies accordingly) which occur during the period of organogenesis
which is up to 8 weeks post fertilization for most organs. E.g. neural tube defect, ventricular septal defect,
polydactyly etc.
Deformation: Distortion of a normally developed structure caused by mechanical forces usually in the latter
half of gestation and most often involving musculo-skeletal tissues. E.g. club foot, torticollis, plagiocephaly
etc.
Disruption: Breakdown of an intrinsically normally developing/ developed tissue due to some disruptive
event such as a mechanical, vascular or infectious insult. E.g. amniotic band sequence.
Dysplasia: Abnormal cellular organization within a tissue, almost always of genetic cause. E.g. skeletal
dysplasias.
A syndrome is a recognized composite pattern of 2 or more anomalies with a common specific aetiology. E.g. Turner
syndrome, fetal phenytoin syndrome etc.
An association is a non-random occurrence of 2 or more anomalies that occur together more frequently than expected
by chance alone, but without a known specific aetiology. E.g. VACTERL (vertebral defects, anal atresia
or stenosis, cardiac defects, tracheo-esophageal fistula, radial defects and renal anomalies, limb defects)
association.
A sequence is a pattern of anomalies resulting from a single primary anomaly or factor E.g. Potter sequence (Primary
anomaly - bilateral renal aplasia/ dysplasia → decreased fetal urine production → severe oligohydramnios →
compressive effects → flattened facies with flattened nose, deformed ears, pulmonary hypoplasia & positional limb
defects).
Approach to a case with dysmorphism
The following step-wise clinical approach should be followed in the assessment and management of an individual with
dysmorphism or congenital malformation(s):
1.
Suspicion of a genetic etiology
2.
Clinical evaluation
history
physical examination
3.
Investigations
4.
Analysis and diagnosis
5.
Confirmation
6.
Intervention:
treatment
counseling
prenatal diagnosis
7.
Surveillance & follow up
Suspicion of a genetic etiology
A genetic aetiology should be suspected in any individual with the following:
Congenital anomalies: at least 1 major/ > 2 minor anomalies.
Growth deficit (short stature/ failure to thrive)
Developmental delay, intellectual disability or developmental regression
Failure to develop secondary sexual characteristics
Abnormal genitalia
Appears ‘different’/ ‘unusual’
History
A detailed history covering the following aspects should be obtained:
Prenatal history:
Teratogenic exposures especially in the first trimester of pregnancy: infections/ medications/ drugs of
abuse/ maternal illness/ radiation exposure
Prenatal complications and antenatal ultrasonographic findings
Perinatal history:
Presentation/ mode/ complications of delivery
Gestational age and condition (Apgar score) at birth
Birth weight, birth length and head circumference; body proportions
Neonatal course:
Feeding and activity
Any adverse events/ complications
Post neonatal:
Physical growth
Developmental milestones
Neurological symptoms especially seizures / visual or hearing deficits/ behavioural phenotype
Other systemic symptoms
Family history:
At least three generation family history / pedigree
History of recurrent pregnancy losses/ infertility
Specific information/ medical records of other affected family members
Consanguinity in parents
Ethnic background
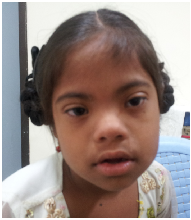
a)
b)
c)
Figure 1: Typical gestalt of some syndromes. a). Down syndrome; b). Cornelia de Lange syndrome; c). Noonan
syndrome.
Physical examination
A thorough clinical examination must be done taking the following aspects into consideration:
General principles:
Thorough head to toe examination to be done.
Measurements to be taken and compared with standard tables/ graphs of age and gender norms.
Both parents and other available family members to be examined for similar or related features.
Clinical photographs to be taken with informed consent of individual/ parent/ guardian: for records,
syndrome search, referral and study of evolution of the phenotype.
Anthropometric measurements:
Height/ length, weight, head circumference
Assessment of proportionality & symmetry:
Upper segment/ lower segment ratio
Arm span
Individual limb segment measurements (in specific cases)
Head to toe assessment: (for exact description of each feature refer to Am J Med Genet A 2009 Jan; 149A (1) &
Aase JM Diagnostic Dysmorphology textbook).
Each body part to be examined carefully from head to feet to look for anomalies
Cranium – size; fontanelles; sutures; shape and symmetry
Scalp hair - colour and texture; distribution; hair whorl patterns; position of anterior and posterior
hairline
Face
overall impression of facial appearance: gestalt e.g. Down syndrome facies, coarse facies, myopathic
facies. See figure 1.
overall shape, symmetry and size of face: triangular/ broad/ round
face to be divided into sections: forehead, midface and oral region
face to be viewed from front and from side
lateral profile better for: depth or height of structures such as nasal bridge, position of mandible
relative to maxilla and midface development
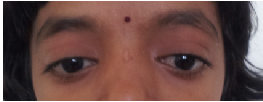
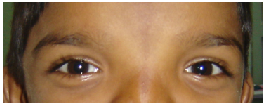
Eyes- eyebrows; palpebral fissure length (short/long); palpebral fissure slant (up/down); epicanthic folds;
eye spacing (use a rough guide of 1:1:1 for ratio of left palpebral fissure length: inner canthal
distance: right palpebral fissure length); palpebral fissure shape; iris colour; pupil shape; cornea/
sclera/ lens; globe position (assessed from lateral view: protuberant vs deep set globes). See figure
2.
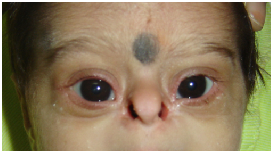
Nose – nasal root; nasal bridge : depressed/prominent/broad; nasal tip: broad/ flattened; columella (the
vertical ridge separating the nostrils): wide/ overhanging; nostrils : patency and position (anteverted); alae
nasi. See figure 3.
Mouth and perioral region - mouth size and shape; upper and lower lip shape and thickness;
gum thickness; philtrum definition and length; jaw position (prognathia/micrognathia); palate
shape
Oral cavity - teeth/ frenulum/ tongue size and morphology
Ears
Ear position
Ear rotation (normally 15 degrees posterior to the vertical plane of the head): anteriorly/ posteriorly
rotated
a)
b)
c)
Figure 3: Dysmorphic findings in the nose. a). Hypoplastic alae nasi; b). Beaked nose; c). Broad bifid tip of
nose.
Position of anus relative to genitalia and patency of anus
Systemic Examination: cardiovascular/ per abdomen/ neurological/ respiratory
Physical features not found as normal or familial traits and which are present in only a few conditions
or are pathognomonic of specific disorders are of more diagnostic help. These are said to be ‘good
handles’ for diagnosis e.g. white forelock of hair which is a good diagnostic clue for Waardenburg
syndrome.
Radiographs
The following radiographic assessment helps in the diagnostic evaluation:
X ray wrist + hand (anteroposterior (AP) view) in cases with short stature: for bone age assessment
Genetic skeletal survey for suspected skeletal dysplasias/ disproportionate short stature:
AP & lateral views of skull
AP & lateral views of spine (cervical to sacrum)
AP view of pelvis with bilateral hip joints
AP view of one hand and one foot
AP view of one upper limb (shoulder to elbow; elbow to wrist)
AP view of one leg (knee to ankle)
Imaging studies
The following imaging modalities may be used in the evaluation:
Neuroimaging:
MRI brain: in presence of neurological deficits/ seizures/ microcephaly or macrocephaly
USG abdomen/ 2D Echo: to look for visceral malformations
Analysis
All clinical and laboratory findings must be analysed together in order to get a diagnosis; all features must
fit into the diagnosis as far as possible
If the condition cannot be diagnosed based on previous experience or existing knowledge, one should take
the help of resources such as dysmorphology databases (e.g. LDDB - London Dysmorphology DataBase and
POSSUM – Pictures of Standard Syndromes and Undiagnosed Malformations), online resources (OMIM –
Online Mendelian Inheritance in Man) and dysmorphology textbooks.
Genetic Testing
The following genetic tests can help in confirming the aetiology in affected cases:
Karyotyping: to be done in cases with:
congenital malformations
prenatal onset growth retardation
disorder of sexual development
developmental delay
history of multiple miscarriages in the family
Fluorescence in situ hybridization (FISH)/ Multiplex ligation - dependent probe amplification (MLPA): when the
phenotype is suggestive of a specific microdeletion syndrome e.g. Di George syndrome (22q microdeletion)/
Angelman syndrome (15q microdeletion)/ Williams syndrome (7q microdeletion)
Metabolic testing: Relevant biochemical investigations should be done if a metabolic etiology is suspected. Metabolic
disorders with dysmorphism include:
Disorders of cholesterol metabolism (e.g. Smith Lemli Opitz syndrome)
Single gene mutation analysis: DNA-based molecular genetic tests to be done when a specific monogenic disorder is
suspected.
Cytogenetic microarray (CMA) study:
Can be done in any case with multiple malformations with or without associated intellectual disability
and without any other identified genetic/ non-genetic cause
CMA scans the entire genome for copy number variations (microdeletions/ microduplications)
Intervention
Appropriate medical/ surgical management wherever feasible: eg. surgical correction of cardiac defect,
correction of hearing deficit etc.
Genetic counseling
Prenatal diagnosis wherever feasible
Genetic Counseling
Deformations/ disruptions have low risk of recurrence (but can recur if the causative intrauterine
environmental factor persists or recurs in the next pregnancy).
Denovo chromosomal abnormalities and microdeletions have a risk of recurrence of < 1%
In single gene disorders, risk of recurrence will vary according to the mode of inheritance: autosomal dominant
(50% in sibs and offspring if inherited and nil in sibs if de novo)/ autosomal recessive (25% in sibs)/ X-linked
(50% in male sibs)
Prenatal Diagnosis
Targeted mutation analysis/ chromosomal analysis/ metabolic testing in fetal tissue depending upon
diagnosis of proband: Chorionic villus sample/ amniotic fluid/ pre-implantation genetic diagnosis
Fetal anomaly scan to look for the same/ associated malformations
3D/ 4D USG for better visualisation of the facial profile/ external dysmorphisms
Fetal echocardiogram for detecting fetal cardiac anomalies
Limitations of scan based prenatal diagnosis:
may not be able to detect certain malformations especially gut anomalies such as malrotation and lower
GI obstruction
cannot determine intellectual status
cannot pick up some features e.g. microcephaly/ lissencephaly until late gestation
Follow up
To assess growth & development
To study course of the disease
To monitor for known/ anticipated associated complications
To offer newly available diagnostic tests
To offer newly available therapeutic options
Sometimes phenotype evolves with age and reassessment at a later age in an undiagnosed case might make
diagnosis clear
To discuss reproductive risks.
Resources for reference
Books:
Aase JM. Diagnostic dysmorphology. 1990. Springer.
Hennekam R, Allanson J, and Krantz I. Gorlin’s Syndromes of the Head and Neck. Fifth edition; 2010.
Oxford University Press.
Hall JG, Allanson JE, Gripp KW, Slavotinek AM. Handbook of Normal Physical Measurements. Second
edition; 2007. Oxford University Press.
Jones KL. Smith’s Recognizable Patterns of Human Malformation. Sixth edition; 2005. Elsevier.
Langman’s Medical Embryology. Sadler TW. Twelfth edition; 2012. Lippincott Williams & Wilkins.
Stevenson RE, Hall JG. Human Malformations and Related Anomalies. Second edition; 2006. Oxford
University Press.
 a)
a) 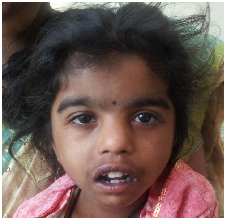 b)
b) 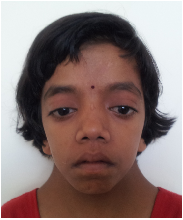 c)
c)
 a)
a)
 b)
b) 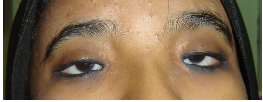 c)
c)
 d)
d)
 a)
a) 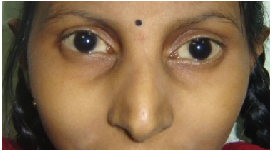 b)
b) 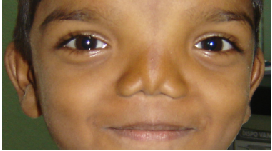 c)
c)