| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
Clinical Vignette
| Table 1:
Summary of phenotypic features in fetuses with Wolf-Hirschhorn syndrome (Xing et al., 2018; Zhen et al., 2018; Chen et al., 2020; Siminino et al., 2022) |
||
|
Clinical feature |
Number (%) out of 83 fetuses |
Fetus in this report |
| Fetal growth restriction | 68/83 (81.92%) |
+ |
| Facial anomalies | 58/83(69.87%) |
+ |
| Microcephaly | 16/83(19.27%) |
- |
| Abdominal anomalies | 10/83 (12.04%) |
- |
| Cardiac anomalies | 25/83 (30.12%) |
+ |
| Skeletal anomalies | 17/83 (20.48%) |
+ |
| Cerebral anomalies | 19/83 (22.89%) |
- |
| Pulmonary malformation | 12/83 (14.45%) |
- |
| Cystic hygroma | 8/83 (9.63%) |
- |
| Increased nuchal translucency or nuchal fold thickness | 18/83(21.68%) |
- |
Discussion
WHS, chromosome 4p monosomy, is a contiguous gene deletion syndrome. The unique features of 4p deletion syndrome include growth retardation, ‘Greek warrior helmet’ facial dysmorphism, intellectual disability, facial clefts, cardiac septal defects, corpus callosum agenesis, kidney abnormalities, feet malformations, congenital diaphragmatic hernia, increased nuchal fold thickening, and cystic hygroma.
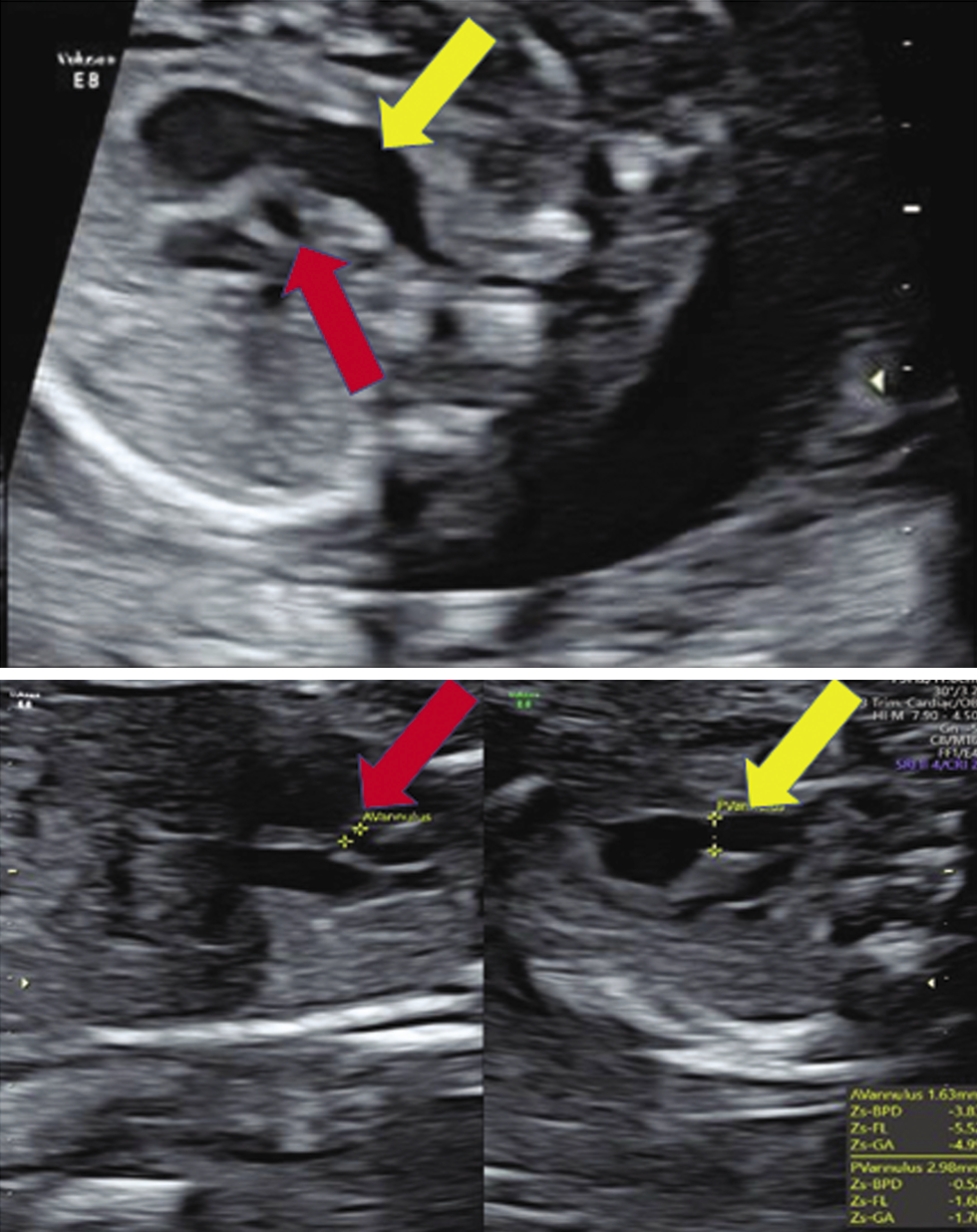
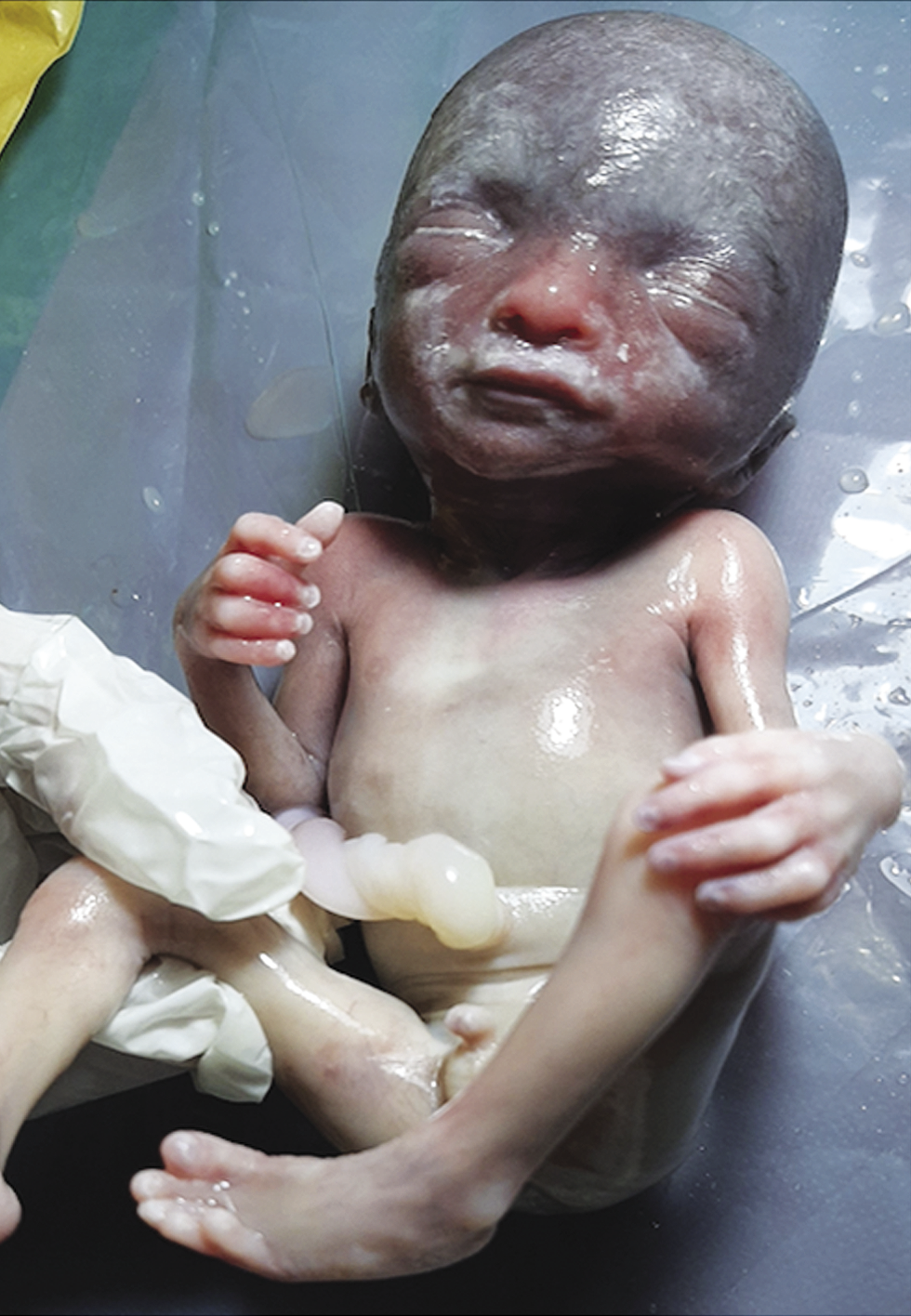
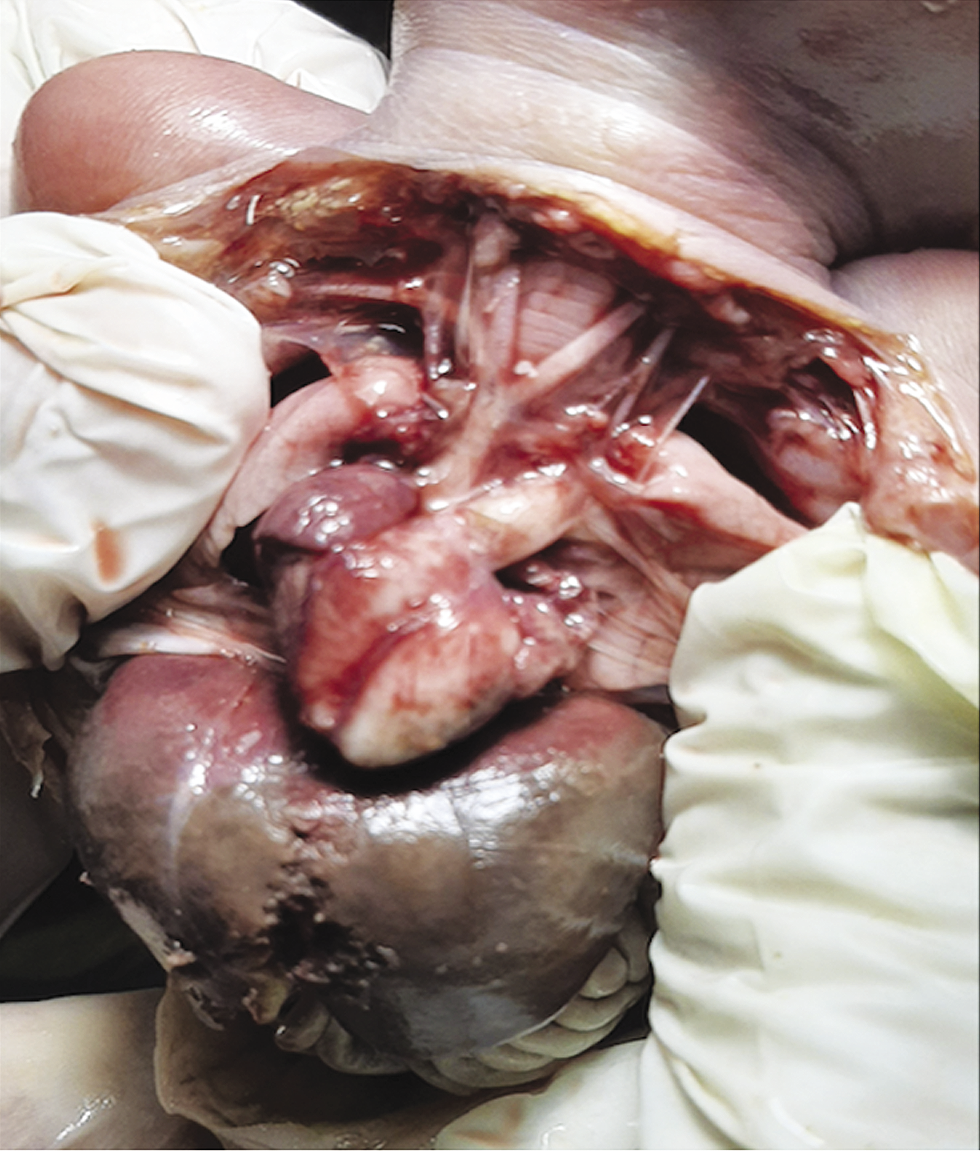
The fetus in this study presented with mild fetal growth restriction (FGR), aortic atresia, pulmonary artery dilatation, and cerebellar hypoplasia on ultrasound. Fetal autopsy findings confirmed the same. Facial features were subtle, like prominent glabella and hypertelorism. In addition, both feet had an abnormal shape. Narrow aorta and dilated pulmonary artery are novel findings not reported in both the prenatal and postnatal series reported earlier. Xing et al. reviewed 37 cases of WHS reported in the literature along with their ten cases and reported that the standard sonographic features of WHS include intrauterine growth retardation (IUGR) (97.7%), typical facial appearance (82.9%), renal hypoplasia (36.2%), cardiac malformation (29.8%), cleft lip and palate (25.5%), cerebral abnormalities (25.5%), skeletal anomalies (21.3%) and increased nuchal translucency/nuchal fold thickness (19%) (Xing et al., 2018). In Table 1, we have compared about 83 fetuses reported in world literature (Simonini et al., 2022). The two most common features are FGR (fetal growth restriction) (81.92%) and typical facial anomalies (69.87%). (Xing et al., 2018; Zhen et al., 2018; Chen et al., 2020; Simonini et al.,2022). In the fetus we report, growth was less than 1st centile, however, typical facial features were not picked up on the ultrasound and we feel that it is difficult to detect subtle facial features of this syndrome by ultrasound without high index of clinical suspicion of this syndrome.
Cardiac malformations in WHS cases have been reported with a breakpoint between 4p15.2 and 4p16.3. Only three prenatal cases with larger 4p deletions have been reported. Verloes et al. reported a fetus with 4p14 deletion with atrial septal defect (ASD), tricuspid valve hypoplasia, pulmonic valve atresia, right ventricular hypoplasia, and aneurysmal dilation of the ascending aorta (Verloes et al.,1995). von Elten et al. reported a fetus with hypoplastic left heart syndrome (HLHS) (von Elten et al., 2013). The fetus reported by Xing et al. had a 23.4 Mb deletion at 4p15.2 and had a complex heart malformation including interruption of the aortic arch, ventricular septal defect (VSD), and pulmonary hypertension (Xing et al., 2018). Postnatal studies mainly discuss genotype- phenotype in the context of facial features, seizures, and growth delay and not based on cardiac anomalies.
In our case study, the fetus had a 17.3 Mb deletion at 4p16.3. It is a moderate size deletion, and maximum cases of this syndrome reported in literature have moderate size deletions. Patients with deletion <5 Mb cannot be picked up on routine karyotype. Chromosomal microarray has higher resolution compared to the karyotype and can detect sub-microscopic deletions upto 20 kb, and hence should be offered in all cases of fetal ultrasound anomalies and FGR.
Deletion of WHSC1, (abbreviated later as NSD2 gene) (OMIM *602952), can disrupt the regulation of several genes resulting in WHS features (Meckkawy et al., 2021) De novo loss-of-function variants in NSD2 gene were recently reported in patients with atypical WHS and in developmental delay, congenital cardiac defects and autism (Bockzeck et al., 2018). Nimura et al. hypothesized that NSD2 gene might play a role in modulating the cardiac transcriptional network through collaboration with NKX2.5 gene (Nimura et al., 2009).
The recurrence risk is negligible in de novo/ sporadic cases. However, in cases where WHS is due to balanced translocation in parents, recurrence risk could be 30-40% and in such cases prenatal diagnosis can be done by chorionic villus sampling or amniocentesis during pregnancy.
Conclusions
Our report highlights novel findings like narrow ascending aorta and dilated pulmonary artery associated with fetal WHS, and further such studies may provide insight into correlation between the genotype and cardiac phenotype of WHS.
References
1. Battaglia A, et al. Wolf- Hirschhorn (4p-) syndrome. Adv Pediatr. 2001; 48:75 – 113.
2. Battaglia A, et al. Update on the clinical features and natural history of Wolf-Hirschhorn
(4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet C Semin Med Genet. 2008;148C:246 e51.
3. Boczek NJ, et al. Developmental delay and failure to thrive associated with a loss-of-function variant in WHSC1 (NSD2). Am J Med Genet A. 2018; 176: 2798–2802.
4. Chen CP, et al. Wolf-Hirschhorn syndrome: Prenatal diagnosis and molecular cytogenetic characterization of a de novo distal deletion of 4p (4p16.1 pter) in a fetus with facial cleft and preaxial polydactyly. Taiwan J Obstet Gynecol. 2020;59:425-431.
5. Hirschhorn K, et al. Deletion of short arms of chromosome 4-5 in a child with defects of midline fusion. Humangenetik. 1965;1:479-82.
6. Mekkawy MK, et al. Clinical and genetic characterization of ten Egyptian patients with Wolf-Hirschhorn syndrome and review of literature. Mol Genet Genomic Med. 2021;9:e1546.
7. Nimura K, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature. 2009; 460: 287–291.
8. Schneider C, et al. Development of Z-scores for fetal cardiac dimensions from echocardiography. Ultrasound Obstet Gynecol. 2005;26:599-605
9. Simonini C, et al. Prenatal sonographic findings in confirmed cases of Wolf-Hirschhorn syndrome. BMC Pregnancy Childbirth. 2022;22:327.
10. Verloes A, et al. Prenatal diagnosis of cystic hygroma and chorioangioma in the Wolf-Hirschhorn syndrome. Prenat Diagn. 1991;11:129-132.
11. von Elten K,et al. A case of Wolf–Hirschhorn syndrome and hypoplastic left heart syndrome. Pediatr Cardiol. 2013;34:1244–1246.
12. Wolf U, et al. Defizien an den kurzen Armen eines Chromosoms Nr 4. Humangenetik. 1965;1:397 e413.
13. Xing Y, et al. Prenatal diagnosis of Wolf-Hirschhorn syndrome: from ultrasound findings, diagnostic technology to genetic counseling. Arch Gynecol Obstet. 2018;298:289-295.
14. Zhen L, et al. Prenatal diagnosis of Wolf-Hirschhorn syndrome: Ultrasonography and molecular karyotyping results. Eur J Obstet Gynecol Reprod Biol. 2018;225:19-21.
15. Zollino M, et al. Genotype-phenotype correlations and clinical diagnostic criteria in Wolf-Hirschhorn syndrome. Am J Med Genet. 2000;94:254-261.
16. Zollino M, et al. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: Genotype–phenotype correlation analysis of 80 patients and literature review. Am J Med Genet Part C Semin Med Genet. 2008; 148C: 257–269.
| Abstract | Download PDF |