1 Institute of Medical Genetics and Genomics, Sir Ganga Ram Hospital, New Delhi, India 2 Child Development Clinic, Institute of Child Health, Department of Pediatrics, Sir Ganga Ram Hospital, New Delhi, India 3 Sir Ganga Ram Hospital, New Delhi, India
4 Department of Pediatrics, Sir Ganga Ram Hospital, New Delhi, India
Correspondence to: Dr Veronica Arora Email: veronicaarora@gmail.com
Abstract
Glucose transporter type 1 (GLUT1) deficiency is a treatable neurometabolic disorder caused due to defect in the facilitative glucose transporter, GLUT1, present in the brain. Variants in SLC2A1 are known to result in GLUT1 deficiency syndrome. We report a 6-year-old male who presented with global developmental delay and epilepsy. Whole exome sequencing identified a novel heterozygous likely pathogenic variant, c.1279-8_1288del in exon 10 of SLC2A1. This also unmasked the diagnosis of his mother who also had the same variant with milder. Diagnosis allowed appropriate management of both the proband and his mother, highlighting the importance of a systematic approach for diagnosis of genetic disorders.
Keywords: Global developmental delay, epilepsy, SLC2A1, GLUT1 deficiency syndrome
Introduction
SLC2A1 (OMIM* 138140), located on chromosome 1p34.2, encodes GLUT1 transporter which is the major glucose transporter in the brain, placenta, and erythrocytes. GLUT1 deficiency syndrome (OMIM #606777) is a treatable neurological disorder characterized by early-onset seizures, developmental delay, spasticity, and motor incoordination. Those affected might show delayed cognition ranging from learning disability to severe forms of intellectual disability (Wang et al., 2018). The disorder is also characterized by low cerebrospinal fluid (CSF) glucose concentration (<40mg/dl), a condition termed hypoglycorrhachia (Wang et al., 2018).
We describe a 6-year-old male child, born to a non-consanguineous couple, with global developmental delay and recurrent seizures with GLUT1 deficiency syndrome-1. The same disorder was identified in his mother who had mild intellectual disability and epilepsy.Both were started on a ketogenic diet and showed improvement in symptoms and wearing down on anti-seizures medications.
Clinical details
A 6-year-old male child presented with complaints of global developmental delay and recurrent seizures (Figure 1). He was the first born child of non-consanguineous parents. He had an unremarkable antenatal, perinatal, and postnatal period. The first episode of seizures at the age of 2 months was treated with phenobarbital but he continued to have recurrent seizures. He received multiple anti-seizure medications including levetiracetam, sodium valproate, clonazepam, and oxcarbazepine as per his clinical seizure semiology and electroencephalogram (EEG) findings. However, generalized, and focal seizures continued to occur. He developed spastic quadriparesis and remained non-ambulatory. There was a history of aggressive behavior with delayed speech. Bowel and bladder control had not been attained. Feeding and sleep were normal. At presentation, he had severe global developmental delay and abnormal posture with no concerns for hearing and vision. His mother had seizures and mild intellectual disability. She has had two episodes of seizures which were managed on anti-seizure medications- valproate and levetiracetam.
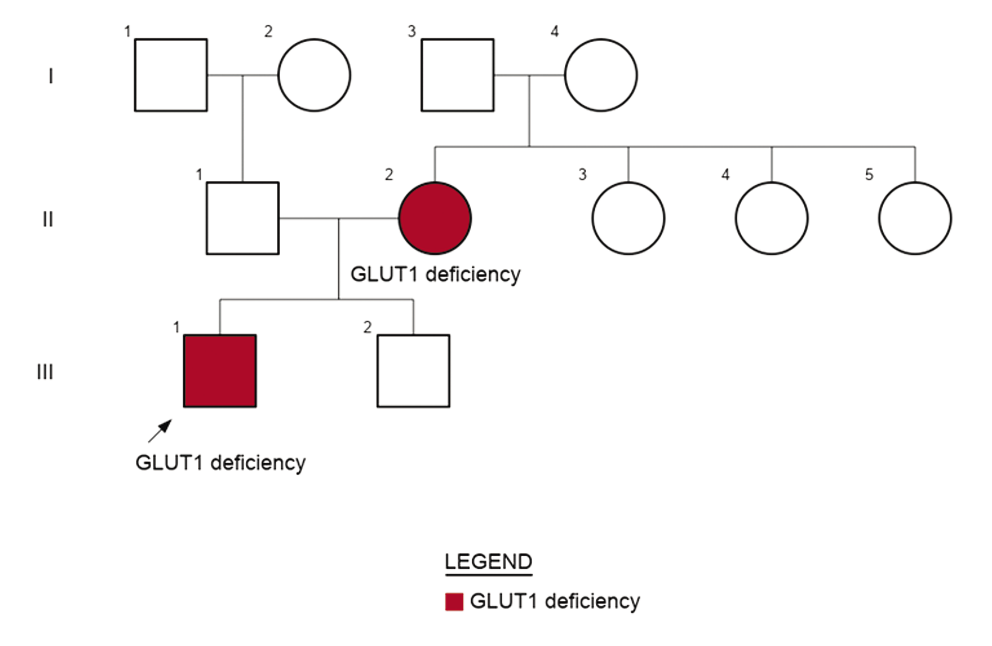
Figure 1: Three generation pedigree showing autosomal dominant inheritance of GLUT1 deficiency syndrome-1 from mildly affected mother to proband.
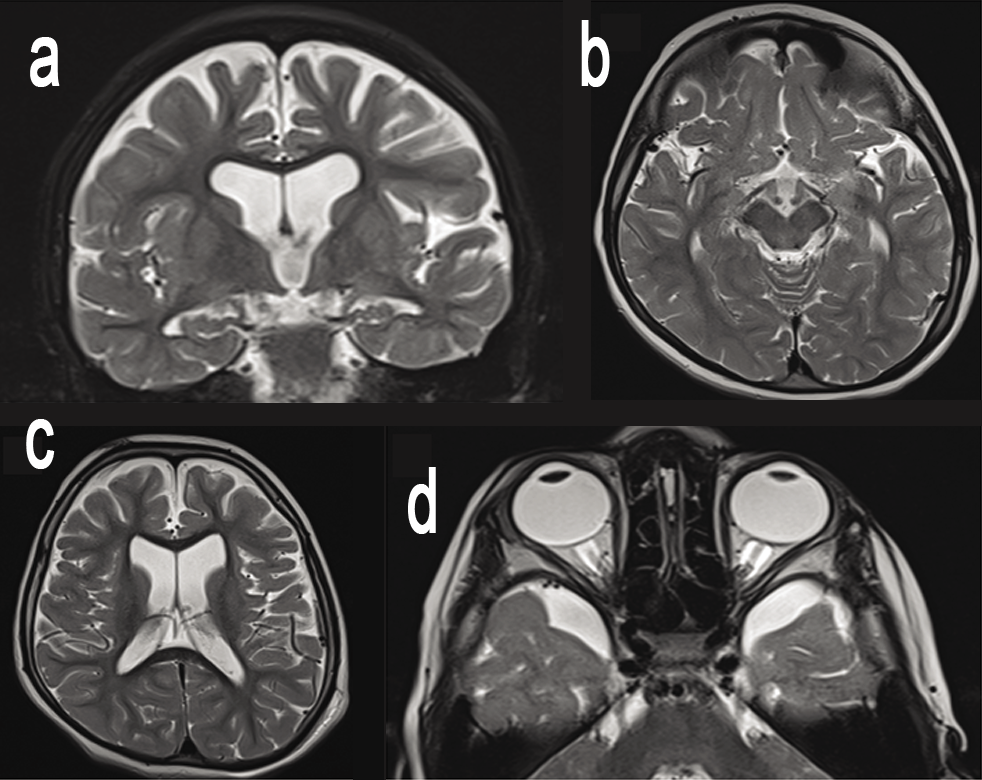
On examination, he was normocephalic with a head circumference of 51cm, height of 110cm (-0.21 SD) and weight 18.5kg (-0.10SD). No facial dysmorphism was noted. He was able to comprehend simple commands like when asked to point towards objects. He could recognize his parents but could not speak specific words. Nervous system examination showed severe spasticity in upper and lower limbs with brisk reflexes. Electroencephalography was suggestive of severe degree of diffuse encephalopathy. Magnetic resonance imaging of brain showed cerebral atrophy with prominent perioptic nerve sheaths (Figure 2). Basic metabolic workup including ammonia, lactate, urine ketones, tandem mass spectrometry (TMS), and urine gas chromatography mass spectrometry (GCMS) were normal.
Figure 2: MRI Brain images of the proband showing cerebral atrophy (a,c), prominent perisylvian fissures (b) and prominent perioptic nerve sheaths (d).
In view of recurrent seizures and significant family history, whole exome sequencing was done, which revealed a novel likely pathogenic heterozygous variation c.1279-8_1288del in exon 10 of SLC2A1, confirming the diagnosis of early-onset GLUT1 deficiency syndrome-1. The variant is an18bp deletion beginning 8bp upstream from the start of exon 10 of SLC2A1. The splice junction loss variant NM_006516.4(SLC2A1):c.12798_1288 del mutates a splice-acceptor sequence resulting in the loss of an acceptor splice site for the clinically relevant transcript. Loss-of-function (LOF) is a known mechanism of disease (gene has 127 pathogenic LOF variants and gnomAD LOF Observed/Expected = 0.0509 is less than 0.755). There are 3 downstream pathogenic loss-of-function variants, with the furthest variant being 42 residues downstream of this variant. This indicates that the region is critical to protein function. The gene SLC2A1 has a low loss-of-function observed/expected upper bound fraction (LOEUF) of 0.24, indicating that the gene is less tolerant to loss-of-function variants. This variant has been classified as likely pathogenic based on the ACMG/AMP criteria, PVS1 [Null variant (intronic within ±2 of splice site) in SLC2A1 where LOF is a known mechanism of disease] and PM2 (absent in population datasets) (Richards et al., 2015). Sanger sequencing confirmed the heterozygous state of the variant in the mother of the proband, thus confirming the variant to be a familial variant causative of disease (Figure 3). Whether the variant in proband’s mother was inherited or sporadic could not be confirmed as her parents were not available for testing.
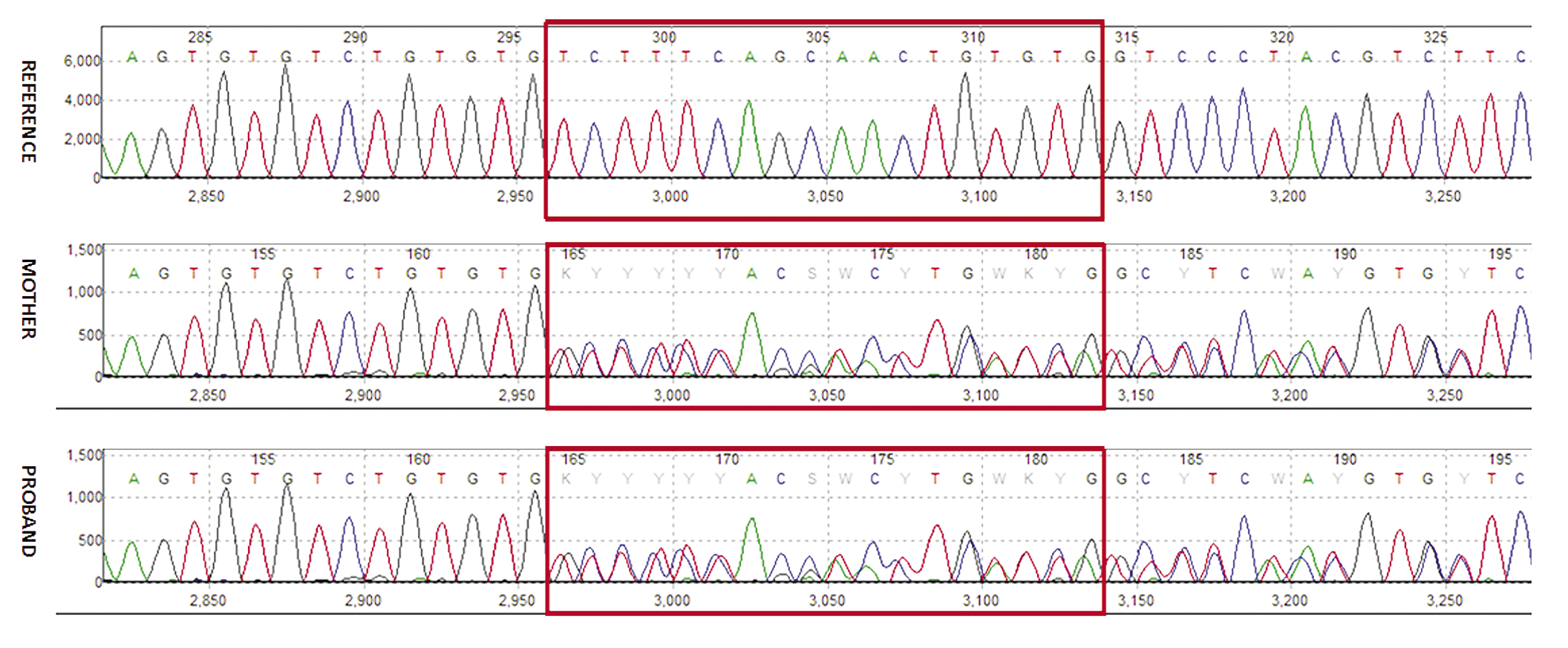
Figure 3: Sanger sequence chromatograms demonstrate the c.1279_1288del variant in exon 10 of SLC2A1 in heterozygous form in proband and his mother.
CSF glucose studies were performed for the proband as confirmation and found to be low (38mg/dL). Treatment with a ketogenic diet was started immediately with carnitine supplementation which resulted in significant improvement in seizure and motor symptoms. His anti-seizure medications including sodium valproate were weaned off gradually. He is now seizure-free with a marked catch-up in his developmental milestones. His mother was evaluated and managed. She has been seizure free for 4 years now.
This shows the importance of genetic testing in this family and how a diagnosis was made possible for our proband’s mother who had a milder phenotype. Reproductive counseling was offered to the couple subsequently. Both the child and the mother are under follow-up.
Discussion
GLUT1 deficiency syndrome is a well-characterized neurometabolic disorder that has a continuum of phenotypic spectrum. The phenotype of the disorder ranges from the most severe “classical” form to mild presentation in affected children (Isamayilova et al., 2018). The classic phenotype includes early-onset seizures before the age of 4 months of age (unresponsive to anti-epileptic drugs), delayed development, motor incoordination, spasticity, and acquired microcephaly. Cognitive impairment can include learning disabilities to severe intellectual delay in some affected individuals (some affected with normal IQ) while some affected individuals may also present with confusion, lethargy, and ataxia (Wang et al., 2018). The type of seizures varies in semiology and frequency. Mostly, these paroxysmal episodes are observed to worsen under stressful situations, during fasting and infections (Wang et al., 2018).
Most cases of GLUT1 deficiency syndrome are inherited in an autosomal dominant mode while a few cases of autosomal recessive transmission have been observed (Brockmann et al., 2001). Nearly 90% of the diagnosed cases are caused due to de novo pathogenic/likely pathogenic variants with unaffected parents, whereas 10% of affected children inherit the pathogenicvariant from their affected parents who might be mildly affected (Wang et al., 2018). The proband we have described has inherited the variant from his mildly affected mother.
The deficiency is caused due to defect in the GLUT1 transporter which is a facilitative glucose transporter of the GLUT family of proteins (Wang et al., 2018). It is responsible for the transfer of glucose across the blood-brain barrier which is the principal source of energy for the brain. Pathogenic/likely pathogenic variants in SLC2A1 can cause a defect in GLUT1 transporter impairing the transport of glucose in the brain and thus causing hypoglycorrhachia (CSF glucose <40mg/dl) which results in seizures, encephalopathy with motor and developmental delay in affected children (Wang et al., 2018). The metabolic rate of glucose in the brain is low in fetal life and increases in early infancy and childhood which results in clinical manifestation of the disorder in early infancy (Wang et al., 2018). The persistent hypoglycorrhachia in our patient explains the severe intellectual disability and seizures.
Ketogenic diet is the primary treatment management offered to the affected children. Ketones are used by the brain as an alternative fuel source. It is well tolerated and is a proven treatment to reduce seizures and movement disorders while it has no to low effect on the cognitive development of the child (Wang et al., 2018). Treatment with ketogenic diet is most beneficial in early childhood as it marks a crucial time for brain development (Klepper et al., 2001). In our patient, ketogenic diet along with carnitine supplementation was started at diagnosis, but due to diagnostic delay, complete reversal of the already existing brain injury was not possible. There was a decrease in the frequency of seizures. He has been seizure-free since the beginning of treatment. The ketogenic diet was also started in the mother and over a period of 6 months resulted in better engagement in day-to-day activities and improvement in self-care as reported by family members.
In conclusion, GLUT1 deficiency should be suspected in children with dystonia, spasticity and developmental delay as an early diagnosis which can lead to better cognitive and behavioral outcomes.
References
1. Brockmann K, et al. Autosomal dominant Glut-1 deficiency syndrome and familial epilepsy. Ann Neurol. 2001; 4:476–485.
2. Ismayilova N, et al. GLUT-1 deficiency presenting with seizures and reversible leukoencephalopathy on MRI imaging. Eur J Paediatr Neurol. 2018; 6:1161–1164.
3. Klepper J, et al. Autosomal dominant transmission of GLUT1 deficiency. Hum Mol Genet. 2001; 1:63–68.
4. Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405-424.
5. Wang D, et al. Glucose transporter type 1 deficiency syndrome. GeneReviews®. 2018; University of Washington, Seattle.
Acknowledgements: The authors thank the patient and his family for their participation.
Conflicts of interest: There are no conflicts of interest.