| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
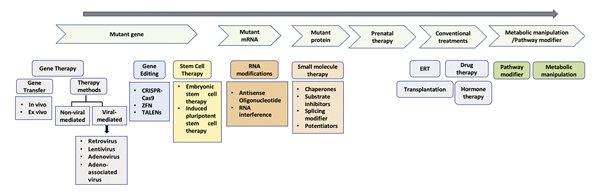
GeNeViSTA
|
Disorder |
Enzyme |
Dietary |
Additional therapeutic options
|
|
Disorders of Amino Acid Metabolism |
|||
|
Phenylketonuria |
Phenylalanine hydroxylase (PAH) deficiency |
Phenylalanine-restricted diet |
Tetrahydrobiopterin, large neutral amino acids, phenylalanine ammonia lyase enzyme therapy(emerging treatment) |
|
Tetrahydro-biopterin (BH4) deficiency |
Six disorders of disturbance of BH4 biosynthesis or recycling |
Phenylalanine-restricted diet |
Sapropterin dihydrochloride, L-DOPA, 5 hydroxytryptophan, folinic acid |
|
Tyrosinemia I |
Fumaryl Acetoacetate hydrolase deficiency
|
Phenylalanine and tyrosine-restricted diet |
Nitisinone (NTBC) |
| Classical homocystinuria | Cystathionine beta synthase deficiency | Methionine-restricted diet | Pyridoxine (B6), vitamin B12, folic acid, betaine |
| Glutaric acidemia type 1 | Glutaryl CoA dehydrogenase deficiency | Low protein, lysine-free, tryptophan-reduced diet | Riboflavin, L-carnitine supplementation |
|
Propionic acidemia and methylmalonic acidemia |
Propionyl CoA carboxylase and methyl malonyl CoA-mutase deficiency |
Methionine, isoleucine, threonine, and valine-restricted diet |
Biotin, L-carnitine and vitamin B12 supplementation, hemodialysis, and hemofiltration |
|
Maple syrup urine disease |
Branched-chain alpha-keto acid dehydrogenasecomplex deficiency |
Dietary restriction of branched-chain amino acids |
High dose thiamine |
|
Urea cycle disorders |
Deficiency of enzymes of the urea cycle (eight enzymes) |
Protein-restricted diet except in type 2 citrullinemia where lactose-free, MCT-enriched diet is recommended |
Sodium benzoate, phenylacetate, L-arginine (except in arginase deficiency), L-citrulline, carbamylglutamate |
|
GLUT1 deficiency |
Glucose transport 1 deficiency |
Ketogenic diet, avoid valproic acid and carbohydrate sugars |
|
|
Glycogen storage disorder |
Glucose 6-phosphatase deficiency |
Frequent meals with complex carbohydrates; limit simple sugars in diet |
Uncooked cornstarch |
|
Fructose 1,6 bisphosphatase deficiency |
Fructose 1,6 Bisphosphatase deficiency |
Avoid fasting; limit use of fructose, sucrose and sorbitol |
- |
|
Disorders of Lipid Metabolism |
|||
|
Very long-chain acyl CoA dehydrogenase deficiency |
Very-long-chain acyl-coenzyme A dehydrogenase (VLCAD) deficiency |
Frequent feeds and avoid fasting; dietary mix - 10% natural fat - reduction of long-chain fat,essential fatty acids supplementation
|
Medium chain triglyceride supplementation |
|
Urea cycle disorders |
Deficiency of enzymes of the urea cycle (eight enzymes) |
Protein-restricted diet except in type 2 citrullinemia where lactose-free, MCT-enriched diet is recommended |
Sodium benzoate, phenylacetate, L-arginine (except in arginase deficiency), L-citrulline, carbamylglutamate |
|
Disorders of Carbohydrate Metabolism |
|||
|
Classic galactosemia |
Galactose-1-phosphate uridyl transferase |
Galactose and lactose-free diet |
Calcium and Vitamin D supplementation |
Table 1: Treatment options for common inborn errors of metabolism
(Pyeritz et al., 2021; Barigga et al., 2021)
|
Vitamin |
Disorder |
|
Pyridoxine |
|
|
Cobalamin |
|
|
Biotin |
|
|
Thiamine |
|
|
Riboflavin |
|
|
Folate |
|
|
Vitamin A |
|
|
Vitamin D |
|
|
Vitamin E |
|
Table 2: Vitamin-responsive genetic disorders
|
Disorder |
Deficient enzyme |
Approved ERT |
Trade Name |
Regulatory Approval |
|
Gaucher disease |
Glucocerebrosidase |
Imiglucerase Velaglucerase alfa Taliglucerase alfa |
Cerezyme® VPRIV® Elelyso® |
DCGI & FDA DCGI & FDA FDA |
|
Fabry disease |
Alpha-galactosidase A |
Agalsidase-beta Agalsidase alpha |
Fabrazyme® Replagal® |
DCGI & FDA EMA |
|
MPS-I, Hurler/Scheie |
Alpha-L-iduronidase |
Laronidase |
Aldurazyme® |
FDA |
|
MPS-II (Hunter syndrome) |
Iduronate-2 sulfatase |
Idursulfase, Idursulfase beta |
Elaprase® Hunterase® |
DGCI FDA |
|
MPS-IV A(Morquio syndrome) |
N-acetylgalactosamine-6-sulfatase |
Elosulfase alfa |
Vimizim® |
FDA |
|
MPS-VI (Maroteaux-Lamy syndrome) |
N-acetylgalactosamine-4-sulfatase |
Galsulfase |
Naglazyme® |
FDA |
|
MPS-VII (Sly syndrome) |
Beta-glucuronidase |
Vestronidase alfa |
Mepsevil® |
FDA |
|
Pompe disease |
Acid alfa glucosidase |
Alglucosidase alfa |
Myozyme® |
DGCI |
|
Alpha-mannosidosis |
Alpha-mannosidase |
Velmanase alpha |
Lamzede® |
FDA |
|
Hypophosphatasia |
Tissue non-specific alkaline phosphatase |
Asfotase alpha |
Strensiq® |
FDA |
|
Lysosomal acid lipase deficiency |
Lysosomal acid lipase |
Sebelipase alfa |
Kanuma® |
FDA |
|
Adenosine deaminase deficiency |
Adenosine deaminase |
Pegademase bovine Elapegademase-lvlr |
Adagen® Revcovi® |
FDA |
|
Phenylketonuria |
Phenylalanine hydroxylase |
Pegvaliase |
Palynziq® |
FDA |
|
Neuronal ceroid lipofuscinosis type 2 |
Tripeptidyl peptidase 1 |
Cerliponase alpha |
Brineura® |
FDA |
*MPS – Mucopolysaccharidosis, US FDA – United StatesFood and Drug Administration, EMA- European Medicines Agency, DCGI- Drugs Controller General of India
Table 3: Enzyme Replacement Therapies
(Pogue et al., 2018; Barigga et al., 2021; Maldonado et al., 2021).
|
Therapeutic product |
Approval authority & year |
Disorder |
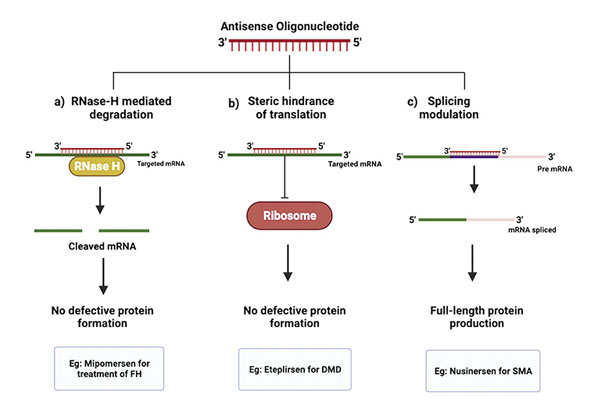
RNA modification & Mechanism of action |
Inclusion criteria |
Effect of therapy |
Approximate treatment cost |
|
Exondys 51 (Eteplirsen) |
USA FDA 2016 |
DMD |
Morpholino ASO designed to cause skipping of exon 51 of the dystrophin gene. |
Mutation in DMD gene amenable to exon 51 skipping |
It causes exon skipping resulting in a short protein with greater functionality |
$300,000 annually per patient |
|
Vyondys 53 (Golodirsen) |
US FDA 2019 |
DMD |
Morpholino ASO designed to cause skipping of exon 53 of the dystrophin gene. |
Mutation in DMD gene amenable to exon 53 skipping |
It causes exon skipping resulting in a short protein with greater functionality |
$300,000 annually per patient |
|
Viltepso (Viltolarsen) |
US FDA 2020 |
DMD |
Second approved morpholino ASO designed to cause skipping of exon 53 of the dystrophin gene. |
Mutation in DMD gene amenable to exon 53 skipping |
It causes exon skipping resulting in a short protein with greater functionality |
$1300 for 5ml vial |
|
Amondys 45 (Casimersen) |
US FDA (2021) |
DMD |
Morpholino ASO designed to cause depletion of exon 51 of the dystrophin gene. exon skipping of exon 45 and dystrophin synthesis |
Mutation in DMD gene amenable to Exon 45 skipping |
It causes exon skipping resulting in a short protein with greater functionality |
$1680 for 2ml vial |
|
Spinraza (Nusinersen) |
US FDA 2016 |
SMA type-1 |
ASO which targets intron 7 on the SMN2 hnRNA modulating alternative splicing by increasing inclusion of exon 7 in the final processed RNA |
SMA patients who have at least one copy of the SMN2 gene |
Modify the expression of SMN2 |
$125000 per injection |
|
Kinamro (Mipomersen) |
US FDA 2013 |
Familial hypercholesterolemia (FH) |
ASO that interferes with the synthesis of ApoBresulting in RNase H-mediated disruption of the mRNA molecule |
Variants in the LDLR, APOB, PCSK9 genes |
Reduce the synthesis of ApoB in the hepatocytes |
$6910 for 1ml vial |
|
Tegsedi (Inotersen) |
US FDA 2018 |
Familial amyloid polyneuropathy (FAP) |
ASO that causes degradation of mutant and wild-type TTR mRNA through binding to the TTR mRNA |
All FAP diagnosed patients |
Reduction of serum TTR protein and TTR protein deposits in tissues |
$420,000 annually per patient |
|
Onpattro (Patisiran) |
US FDA 2018 |
Familial amyloid polyneuropathy (FAP) |
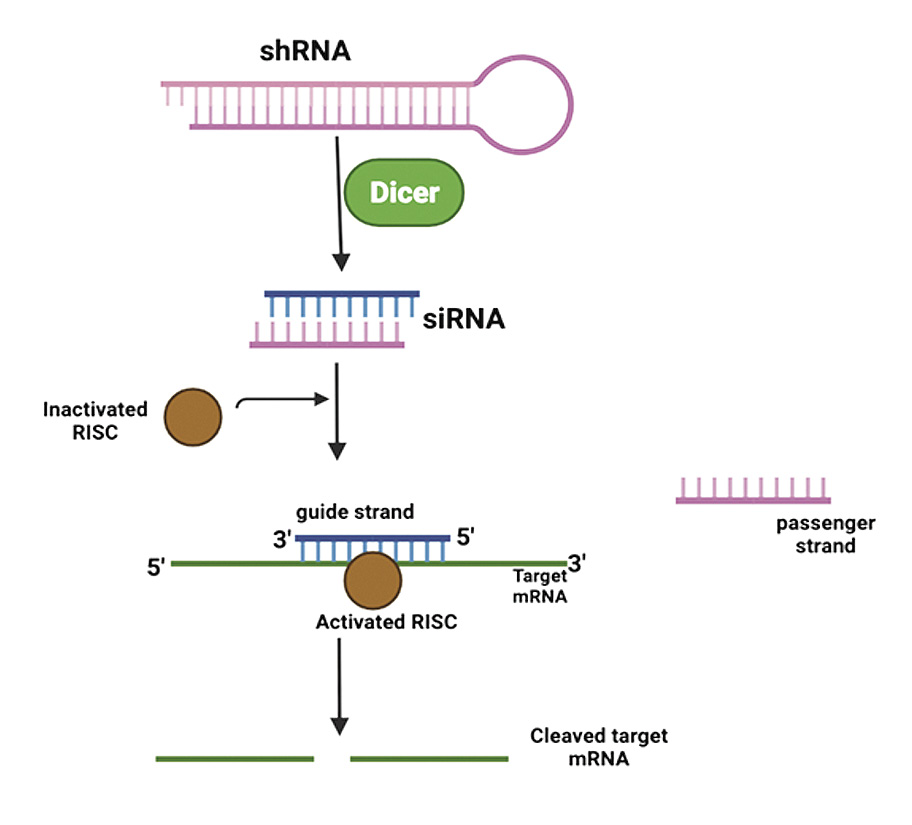
Lipid nanoparticle containing an RNAi targeting the transthyretin mRNA |
All FAP diagnosed patients |
Results in a reduction of mutant protein |
$345000 per 2 mg/ml |
|
Givlaari (Givosiran) |
US FDA 2019 |
Acute hepatic porphyria (AHP) |
Aminolevulinate synthase 1 (ALAS1) directed RNAi |
All AHP diagnosed adult patients |
Degradation of ALSA1 mRNA in hepatocytes reducing its elevated levels in liver |
$575000 per year per patient |
|
Oxlumo (Lumasiran) |
US FDA 2020 |
Primary hyperoxaluria type 1 (PH1) |
RNAi that reduces the levels of glycolate oxidase enzyme by targeting glycolate oxidase encoding mRNA |
All PH1 diagnosed patients |
Reduction of glycolate oxidase levels by silencing of gene encoding glycolate oxidase |
$493000 per year per patient |
Table 4: Therapeutic products based on RNA modification therapy
(O'Connor et al., 2006; Turnpenny et al., 2017; Shahryari et al., 2019).
US FDA – United States Food and Drug Administration, DMD – Duchenne muscular dystrophy, SMA – spinal muscular atrophy
|
Vector |
Retrovirus |
Lentivirus |
Adenovirus |
Adeno-associated virus |
|
Viral genome |
RNA |
RNA |
dsDNA |
ssDNA |
|
Transfection capacity |
<8kb |
8-10kb |
8-30kb |
4.5-8kb |
|
Genome integration |
Yes |
Yes |
No |
No |
|
Long-term expression |
Yes |
Yes |
No |
Yes |
|
Immune response to vector |
Few |
Few |
Yes |
No |
|
Cell division requirement for target cell |
Yes |
G1 phase |
No |
No |
|
Limitations |
Risk of insertional mutagenesis; only infects dividing cells |
Risk of insertional mutagenesis |
Contains genes involved in the process of malignant transformation, so there is a potential risk of induced malignancy |
Can be activated by any adenovirus infection; causes immune response |
|
Advantages |
Persistent gene transfer in dividing cells |
Can be integrated into non-dividing cells, useful in the treatment of neurological conditions |
Infect a wide variety of cell types, stable, can infect non-dividing cells, they have a greater capacity to infect different tissues |
Infect a wide variety of cell types and nonpathogenic |
|
Treatment for |
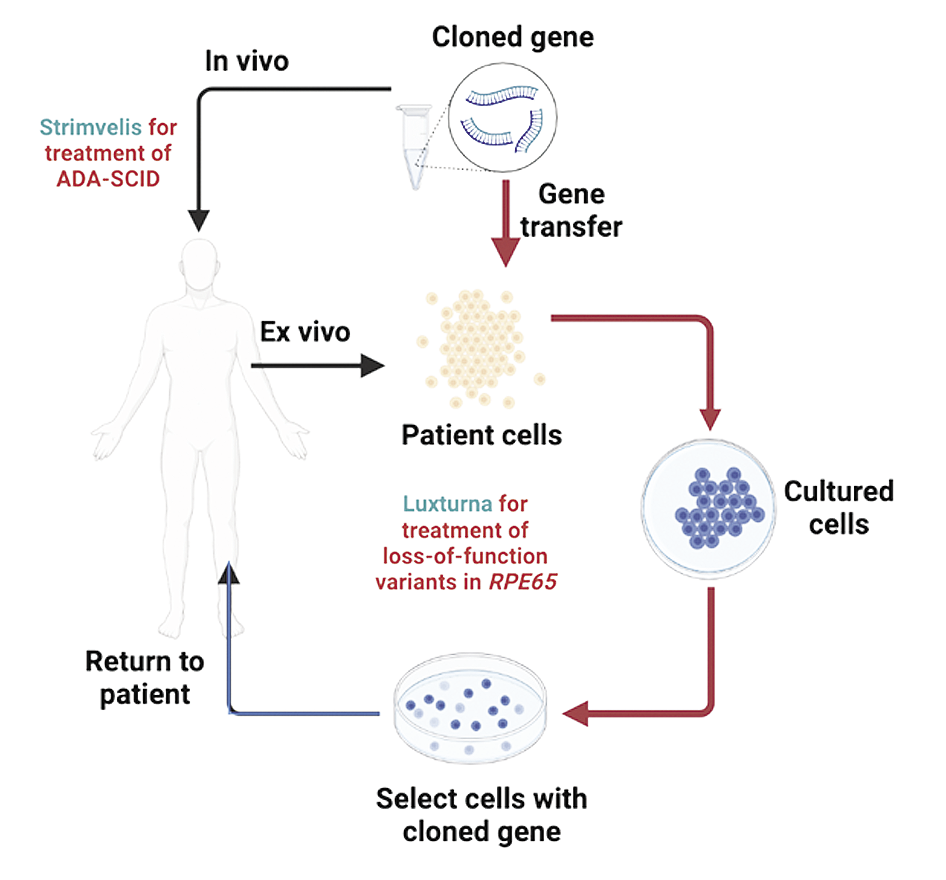
ADA-SCID |
Neurological conditions, beta-thalassemia |
Cystic fibrosis |
Retinal dystrophy caused by bi-allelic loss of function RPE65 mutations |
Table 5: A comparison of viral vectors for gene transfer
(O'Connor et al., 2006; Turnpenny et al., 2017; Maldonado et al., 2021)
|
Product name |
Approval authority |
Disorder |
Vector and mechanism of action |
Limitation |
Price |
|
Luxturna (VoretigeneNeparvovec-rzyl) |
US FDA 2017 |
Retinal dystrophy caused by bi-allelic loss of function RPE65 mutations |
AAV2 carrying a normal copy of the RPE65 gene |
Conjunctival hyperemia, cataract, increased intraocular pressure and retinal tear, holes, and inflammation |
$850,000 per patient, $425,000 per eye |
| Zolgensma (OnasemnogeneAbeparvovec) |
US FDA 2019 |
SMA 1 patients <2 years of age |
Non-replicating recombinant AAV9 containing a functional copy of human SMN1 gene under the control of CMV enhancer/chicken-β-actin-hybrid promoter |
Benefits of the drug in patients with advanced SMA not recorded |
$2.125 million for a one-time treatment |
|
Strimvelis (GSK-2696273) |
EMA 2016 |
ADA-SCID |
Retroviral vector transduced autologous HSC expressing ADA |
HCV infected patients (> 15 IU/ ml nucleic acid test) |
$648000 per patient |
|
Zynteglo (Betibeglogeneautotemcel) |
EMA 2019 |
Beta-thalassemia (transfusion dependent patients aged 12 years or above) |
Lentiviral associated βA(T87Q)-globin gene sequence |
Thrombocytopenia, not suitable for pregnant or breastfeeding women |
$1.8 million for a one-time treatment |
|
RoctavianTM (Valoctocogene roxaparvovec)
|
EC 2022 (conditional approval) |
Severe |
An AAV5- encoding human B domain-deleted factor VIII |
Transient infusion associated reactions and mild to moderate rise in liver enzymes with no long-lasting clinical sequelae |
$2.5 million for one-time treatment |
RNA – ribonucleic acid, DNA – deoxyribonucleic acid, ADA-SCID – adenosine deaminase deficient severe combined immunodeficiency, ss single stranded, ds double stranded.
US FDA – United States Food and Drug Administration, EMA- European Medicines Agency, ADA-SCID – adenosine deaminase deficient severe combined immunodeficiency, SMA – spinal muscular atrophy, AAV – adeno-associated virus, HSC – hematopoietic stem cells, HCV – hepatitis C virus
Table 6: Approved gene therapy products for the treatment of genetic disorders
(Shahryari et al., 2019; Maldonado et al., 2021).
|
Therapeutic Option |
Disorders |
Advantages |
Disadvantages |
|
Dietary management |
Small molecular disorders e.g.,phenylketonuria, tyrosinemia, homocystinuria |
Easy intervention, cheaper treatment approach which can be modified specific to patients' need |
Lifelong therapy; does not correct the gene defect |
|
Enzyme replacement therapy |
Lysosomal storage disorders, phenylketonuria |
Fewer side effects, longer drug history, and wider availability |
Lifelong intravenous therapy; does not cross the blood-brain barrier; immune response to therapy |
|
Substrate reduction therapy |
Gaucher type 1, Mucopolysaccharidosis-III |
Oral therapy, crosses blood-brain barrier, pharmacodynamic response generally complementary to ERTs, does not elicit immune response |
Not widely available |
|
Chaperone therapy |
Fabry disease, cystic fibrosis |
Oral therapy, wide tissue distribution, fewer immunogenicity reactions |
Lifelong intervention and does not cure the disorder |
|
HSCT |
Severe combined immunodeficiency, lysosomal storage disorders, X-linked adrenoleukodystrophy |
Improves the neuronopathic phenotype |
Difficulty to identify a human leukocyte antigen (HLA)-matched donor; procedure regimen related morbidity and mortality; GVHD; limited impact on CNS and skeletal manifestation |
|
Liver transplant |
Urea cycle disorder, tyrosinemia, Wilson disease,maple syrup urine disease |
Provides relief and better lifestyle quality with no dietary restrictions |
Need of donor match and risk of immune rejection |
|
Kidney transplant |
Polycystic kidney disease, primary hyperoxaluria |
Improved lifestyle, prevention of renal failure and recurrent stone formation |
Need of donor match and risk of immune rejection |
|
RNA based therapies |
Spinal muscular atrophy, familial amyloid polyneuropathy, familial hypercholesterolemia |
Personalized treatment, rapid development, target specific |
Expensive treatment which requires regular administration |
|
Gene therapy |
Spinal muscular atrophy, beta-thalassemia, ADA-SCID, retinal dystrophy |
One time dosage which cures the disorder at gene level |
Have some associated side effects, very expensive with many therapies still under research |
|
Prenatal therapy |
Congenital adrenal hyperplasia, heart block, LUTO |
Earliest intervention even before the onset of disease |
Only available for a very few disorders that are diagnosed during fetal life |
|
Hormonal therapy |
Turner syndrome, Prader-Willi syndrome, Noonan syndrome, idiopathic short stature, congenital hypothyroidism |
Easy availability, wide distribution |
Only a management approach, not curative |
Table 7: Summary of different therapeutic approaches
ERT – enzyme replacement therapy, ADA-SCID – adenosine deaminase-deficient severe combined immunodeficiency, HSCT – hematopoietic stem cell transplantation, GVHD – graft versus host disease, CNS – central nervous system, LUTO – lower urinary tract obstruction
| Abstract | Download PDF |