Recurrent Non-immune Hydrops and Epiphyseal Stippling in Fetuses Affected with Infantile Sialic Acid Storage Disease
Sumita Danda1, Manisha Madhai Beck2, Swati Rathore2
1Clinical Genetics Unit, Christian Medical College & Hospital, Vellore, Tamil Nadu, India
2Department of Obstetrics and Gynaecology, Christian Medical College & Hospital, Vellore, Tamil Nadu, India
Correspondence to: Dr Sumita Danda. Email: sumita_danda@hotmail.com
Abstract
This is the report of a woman with two pregnancies, subsequently terminated, for fetuses presenting with non-immune hydrops, and infantogram showing extensive bone stippling. Clinical exome sequencing was carried out in the DNA banked from one of the affected fetuses. This identified a homozygous pathogenic variant; c.110_111delinsGGA (p.Ser37TrpfsTer41) in the SLC17A5 gene,revealing the diagnosis of infantile sialic acid storage disease (ISSD).Stippled epiphyses have been previously reported in-utero as well as in post-delivery radiographs in ISSD. These are the first cases of stippled epiphyses in fetuses affected with ISSD reported from India.
Keywords: Non-immune fetal hydrops; Epiphyseal stippling; Infantile sialic acid storage disease
Introduction
Non-immune hydrops in a fetus is a severe condition which results in excessive accumulation of fluid within the fetal extravascular compartments and body cavities and is the end-stage of a wide variety of disorders. The exact prevalence is unknown and various causes have been implicated such as congenital heart disease, chromosomal disorders, infections, and monogenic disorders; in some cases, it is idiopathic. Next-generation sequencing technology has enabled us to diagnose inborn errors of metabolism antenatally in fetal hydrops cases in the modern era. The prognosis of non-immune hydrops fetalis is generally poor. We describe here a lady where the cause of fetal hydrops was identified to be infantile sialic acid storage disease (ISSD), based on which accurate genetic counselling could be provided and definitive prenatal diagnosis for the subsequent pregnancy could be offered.
Clinical details
A 24-years old woman with history of two previous spontaneous abortions presented to us
in her third pregnancy. She was consanguineously married (fourth-degree consanguinity) and had no other antenatal risk factors. Her anomaly scan, done at18 weeks of gestation, was normal. A follow- up ultrasound scan done at 29 weeks gestation showed moderate fetal ascites, shortlong bones and mild cardiomegaly. Scan findings revealed short long bones with femur length of 39.8 mm (Z score: -3), along with unilateral club foot, possible syndactyly of lower limbs and a bell shaped thorax. Stippling of calcaneum was not detected in the antenatal scan at 29 weeks.
Overall, a guarded prognosis was explained to the couple, and they were counselled regarding genetic evaluation of the fetus. As the lady had two previous spontaneous abortions and the scan in the ongoing pregnancy was abnormal, showing fetal hydrops and other anomalies, the couple was counselled keeping in mind the possibility of chromosomal anomalies. Single gene disorders causing hydrops were also considered. Differential diagnoses included skeletal dysplasias such as achondrogenesis, thanatophoric dysplasia or Jeune asphyxiating thoracic dysplasia and storage disorders such as Gaucher disease, mucopolysaccharidosis, or mucolipidosis. The couple were explained that genetic testing would help in determining the recurrence risk and subsequent planning of pregnancy.
The woman underwent percutaneous umbilical cord blood sampling (PUBS). Due to financial constraints, initially only a karyotype was done. Fetal DNA banking was done so that further evaluation such as exome sequencing could be planned if the karyotype turned out to be normal. Following this the pregnancy was terminated at 29 weeks gestation. A still born baby girl was delivered and an infantogram done showed calcific stippling. The couple, however, did not consent for fetal autopsy.
The fetal karyotype was normal. The parents were counselled, and clinical exome sequencing was planned in the banked fetal DNA. Possibility of chondrodysplasia punctata, both rhizomelic and non-rhizomelic type (Conradi-Hunermann syndrome), and other peroxisomal disorders such as Zellweger syndrome were considered based on the family history of consanguinity, the USG findings, examination of the expelled fetus and the infantogram. Genetic counselling was provided with possibility of monogenic autosomal recessive disorders and clinical exome sequencing in the banked DNA was carried out for diagnostic purposes. This revealed a novel homozygous pathogenic variant c.110_111delinsGGA (p.Ser37TrpfsTer41) in exon 2 ofSLC17A5 (ENST00000355773.5), which is associated with infantile sialic acid storage disease (ISSD). This variant results in frameshift and premature truncation of the protein 41 amino acids downstream to codon 37. This variant has not been reported in population databases such as gnomAD and 1000 Genomes. The in-silico prediction tool MutationTaster2, predicted this variant to be damaging. The reference region is conserved across mammals. No other clinically significant variants were identified. The parents were counselled about the recurrence risk of 25% in every pregnancy. Prenatal diagnosis in the first trimester was offered for subsequent pregnancies. They were counselled about other reproductive
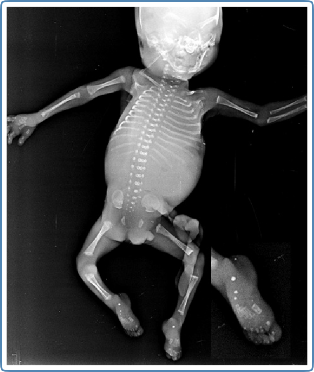
Figure 1: Infantogram showing stippling of hand bones and heels of the secondaffected fetus (calcaneal stippling highlighted in the inset)
options such asin vitro fertilization (IVF) with preimplantation genetic testing (PGT) or use of donor gamete. Extended carrier screeningfor the family was also suggested.
In the subsequent pregnancy, the parents came in the first trimester seeking prenatal diagnosis. Targeted mutation analysis was done in the fetal DNA sample obtained by chorionic villus sampling. This showed an affected fetus due to the presence of the same variant in a homozygous state. The parents were counselled regarding the poor outcome and similar phenotypic manifestations of fetal hydrops; however, they decided to continue the pregnancy. Early anomaly scan, done at 16 weeks, revealed similar fetal phenotype of hydrops with long short bones, as before. In view of scan findings, the couple were offered termination of pregnancy.However, the couple opted for termination only at 20 weeks gestation, when the scan showed gross hydrops and repeat genetic counselling was done,where the poor prognosis was re-emphasized. Infantogram of the expelled fetus revealed epiphyseal stippling (Figure 1).
Discussion
We report here a novel homozygous pathogenic variant in the SLC17A5 gene in two fetuses presenting as non-immune hydrops and short long bones. Infantograms of the terminated fetuses showed epiphyseal stippling. Biallelic pathogenic variants in the SLC17A5 gene cause sialic acid storage disorders which can present as Salla disease (OMIM # 604369), which is mainly reported in the Finnish population, or as a more severe disease known as infantile sialic acid storage disease (OMIM# 269920) which is pan-ethnic. This is an autosomal recessive condition.
SLC17A5 gene codes for the product named sialin (Miyaji et al., 2008). The protein is a vesicular excitatory amino acid transporter (VEAT) which has two functions. In the synaptic vesicles of the central nervous system, sialin causes vesicular storage and subsequently exocytosis of aspartate and glutamate. Whereas in the lysosome, it acts as an H(+) coupled sialic acid exporter(Miyaji et al., 2008).
The founder variant p.Arg39Cys in homozygous state is associated with Salla disease which is characterized by very slowly progressive neurological deterioration. The same variant in compound heterozygous state with other pathogenic variants results in intermediate forms of ISSD. The other variant reported to cause the intermediate type is homozygous p.Lys136Glu, while various other pathogenic variants cause the more severe ISSD phenotype. Allother variants reported till date lead tothe severe phenotype. Among the severe type-causing pathogenic variants, nonsense, missense, frameshift, splice- site and small deletions have been reported.
The infantograms of the terminated fetuses reported here showed calcific stippling; thishighlights the presence of stippled epiphyses with fetal ascites/hydrops as a prenatal and/or early neonatal presentation of severely affected cases of ISSD. This feature along with non-immune hydrops can be confused with other causes of skeletal dysplasia. Stippled epiphyses are seen in the infantogram commonly due to other genetic conditions involving peroxisomal biogenesis disorders such as rhizomelic chondrodyplasia punctata and Zellweger syndrome, or due to maternal factors such as maternal systemic lupus erythematosus (SLE),or warfarin embryopathy. The fetal, maternal, and teratogenic causes of epiphyseal stippling in the perinatal period, are listed in Table 1 (Wainwright&Beighton, 2010; Alrukban & Chitayat, 2018).
Table 1: Summary of the fetal, maternal, and teratogenic causes of perinatal epiphyseal stippling
Etiology | Conditions |
Fetal Factors |
Chromosome abnormalities | |
Peroxisomal disorders | Infantile Refsum disease, Neonatal adrenoleukodystrophy Zellweger syndrome Rhizomelic chondrodysplasia (RCDP1, 2, and 3)
|
Lysosomal storage disorders, | |
Cholesterol synthesis defects | Smith–Lemli–Opitz syndrome Conradi–Hünermann syndrome Greenberg dysplasia Congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD syndrome) Lathosterolosis
|
Abnormal vitamin K metabolism | punctata |
Teratogens |
Infections | Rubella Cytomegalovirus (CMV)
|
Drugs | |
Maternal factors |
Severe malabsorption |
|
Vitamin K deficiency |
|
Maternal autoimmune diseases | |
Froissart and colleagues reported the clinical, morphological, and molecular aspects of the in- utero presentation of SLC17A5-associated ISSD (Froissart et al., 2005). They described the features of eight fetuses in-utero and four neonates, detailing the ultrasonography findings, autopsy findings and molecular testing of the targeted gene. Of the 12 prenatal and perinatal cases, stippled epiphyses were seen in two of the four neonatal cases and six of the eight prenatal cases with the severe ISSD type. Apart from ascites, other features noted in antenatal casesreported by the authors include hepatomegaly, ventriculomegaly, short limbs, short femur, and club feet. Placental hydrops was also found in some cases.
As electron microscopic findings show engorged lysosomes due to abnormal storage of sialic acid, ISSD is classified under the category of lysosomal storage disorders. There is similarity in clinical features namely hydrops, coarse face, and short bones, seen with other lysosomal storage disorders such as mucopolysaccharidosis and mucolipidosis. However, a case series and review of literature by Lemyre et al, mentioned that the features of dysostosis multiplex were mild and corneal clouding was absent(Lemyre et al., 1999).
There is no specific treatment for this condition. The infantile type has a shortened life span and Lemyre et al reported death in early infancy with a mean age of 13.1 months. (Lemyreet al., 1999). Genetic counselling remains an important aspect of management. Prenatal diagnosis remains the mainstay of management in couples with previously affected fetuses or children. This report highlights stippled epiphyses as an important feature of the in-utero presentation of infantile sialic acid storage disease.
References
Alrukban H, Chitayat D. Fetal chondrodysplasia punctata associated with maternal autoimmune diseases: a review. Appl Clin Genet. 2018; 11:31- 44.
Froissart R, et al. Clinical, morphological, and molecular aspects of sialic acid storage disease manifesting in utero. J Med Genet. 2005; 42:829- 836.
Lemyre E, et al. Clinical spectrum of infantile free sialic acid storage disease. Am J Med Genet. 1999; 82:385-391.
Miyaji T, et al. Identification of a vesicular aspartate transporter. Proc Nat Acad Sci. 2008; 105: 11720-11724.
Wainwright H, Beighton P. Lethal epiphyseal stippling in the fetus and neonate;pathological implications. Virchows Arch. 2010; 456:301–308.