| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
GeNeViSTA
Disorder | Gene | Protein/function | Peroxisomes |
and their structure in IHC | |||
i. Group A(Zellweger spectrum disorders) | |||
1. Severe phenotype | PEX1 PEX2 PEX 5 PEX 2,10,12, PEX 13,14 PEX 3,16,19 PEX 6,26 | RING peroxins Peroxisome importome PTS1-linked signaling Peroxisome importome Docking peroxins Peroxisomal membrane pro- teins RING peroxins | Absent |
2. Intermediate phenotype | Absent | ||
3. Milder phenotype | Absent | ||
ii. Group B | |||
1. Rhizomelic chondrodysplasia punc- tate type 1 | PEX7 | PTS2-linked signaling | Enlarged |
2. Rhizomelic chondrodysplasia punc- tata type 5 | PEX5 | PTS1-linked signaling | NA |
NA – Not available
Table 2: Disorders of peroxisome function (Single enzyme deficiency)
Disorder | Gene | Protein/function | Peroxisomes |
and their structure in IHC | |||
i. Etherphospholipid biosynthesis | |||
1. Rhizomelic chondrodysplasia type2 (Dihydroxyacetonephosphate acyltransferase deficiency) | GNPAT | Plasmalogens synthesis | NA |
2. Rhizomelic chondrodysplasia type 3 (Alkyldihydroxyacetone phosphate synthase deficiency) | AGPS | ||
3. Rhizomelic chondrodysplasia type 4 (Fatty acyl-CoA reductase 1 deficiency) | FAR1 | ||
ii.Fatty acid β-oxidation | |||
1. X-linked adrenoleukodystrophy | ABCD1 | VLCFA transporter | Normal |
2. Acyl COA oxidase deficiency | ACOX1 | Peroxisomal β-oxidation | Enlarged |
3. D-Bifunctional protein deficiency | HSD17B4 | Enlarged | |
4. Sterol carrier protein X deficiency | SCP2 | Peroxisomal β-oxidation | NA |
5. Alphamethylacyl COA racemase (AMACR) deficiency | AMACR | NA | |
iii.Fatty acid α-oxidation | |||
1.Refsum disease (Phytanoyl-CoA hydroxylase deficiency) | PHYH | Peroxisomal α-oxidation | NA |
iv.Disorders of Glyoxylate cycle | |||
1.Hyperoxaluria type 1 (Alanine-glyoxylate aminotransfer- ase deficiency) | AGXT | Peroxisomal glyoxylate cycle | Small |
2.Glycolate oxidase deficiency | HAO1 | Glyoxylate metabolism | NA |
v. Bile acid synthesis | |||
1.Bile acid-CoA: amino acid N-acyl- transferase deficiency | BAAT | Peroxisomal acyl-CoA acyl- transferase | NA |
2.Acyl-CoA oxidase 2 deficiency | ACOX2 | Branched-chain acyl CoA oxidase | NA |
3.PMP70 deficiency | ABCD3 | Peroxisomal membrane protein | NA |
vi. H2O2 Metabolism | |||
1.Acatalasemia (Catalase deficiency) | CAT | Antioxidant | Normal |
vii. Lysine catabolism | |||
1.L-lysine oxidation | L-pipecolic acid degradation | NA | |
Abbreviations: IHC-Immunohistochemistry, NA-Not available
4B, but in a few patients with a heterozygous variant in the PEX6 gene, allelic expression imbalance leading to an overrepresentation of a mutant allele and ZSD phenotype has been reported (Falkenberg et al., 2017). Likewise, biallelic pathogenic variants in the FAR1 gene lead to the autosomal recessive peroxisomal fatty acyl-CoA reductase 1 disorder, which is characterized by severe psychomotor retardation during infancy followed by childhood spasticity, but a few patients with a heterozygous pathogenic variant in FAR1 have been reported to have cataracts and spastic paraparesis.
Peroxisome biogenesis disorders also exhibit mosaicism in a few patients. In type 1 mosaicism, normal peroxisomal activity is revealed in fibroblasts with abnormal biochemical profiles. In type 2 mosaicism, with the same genotype,there is a difference in peroxisome morphology in different tissues.
Clinical features of peroxisomal disorders
Peroxisome biogenesis disorders (PBDs)
These are autosomal recessive genetic disorders with an incidence of approximately 1:30,000 to 1:50,000 newborns.The Zellweger spectrum of disorders and rhizomelic chondrodysplasia punctata (RCDP) spectrum are included under this category.
Zellweger spectrum of disorders (ZSDs)
The phenotype of ZSDs usually ranges from severe form to intermediate and milder forms with the typical presentation. Patients with atypical presentation lacking the classical signs and symptoms of ZSDs have also been described. Dysmorphic features in severe ZSDs include the high forehead, large anterior fontanelle, hypoplastic supraorbital ridges, epicanthal folds, corneal clouding, cataract, and broad nasal bridge (Figure 2). Prognosis is guarded with early neonatal or infantile death. Most of the intermediate forms of ZSDs have late childhood deaths. Clues to the diagnosis of Zellweger spectrum disorders in different age groups with the differential diagnosis are depicted in Figure 3. The neonatal phenotype of X-linked adrenoleukodystrophy due to contiguous deletion of the ABCD1 gene which mimics peroxisome biogenesis disorders has also been described.
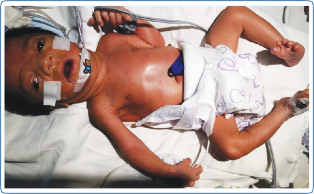
Figure 2: Dysmorphic features in a neonate with the severe Zellweger spectrum disorder
Rhizomelic chondrodysplasia punctata spectrum (RCDP 1 and 5)
The phenotype of RCDP spectrum usually ranges from the severe form who present with midfacial hypoplasia, rhizomelic shortening at birth, to the intermediate form who present in childhood with joint contractures and spastic quadriparesis, and the milder forms who present with mild rhizomelic shortening. Severe phenotype can present either in the prenatal period or in the neonatal period.
Disorders of peroxisome function
Etherphospholipid biosynthesis disorders
RCDP types 2, 3, 4 are included under this category. The clinical features are similar to RCDP 1, 5 and they are differentiated by their genetic etiology. FAR1-related disorder is referred to as RCDP4 by some authors, but this disorder lacks the classical skeletal features of RCDP.
Disorders with impaired fatty acid β-oxidation
X-linked adrenoleukodystrophy (X-ALD): X-ALD is an X-linked recessive disorder. It does not have any clinical features at birth. It can manifest with three different phenotypes. Different forms of X-ALD, their specific clinical features, and their age of presentation are depicted in Figure 4. Some female carriers can present with adrenomyeloneuropathy. Alpha methylacyl-CoA racemase (AMACR) deficiency, acyl-CoA oxidase 1 (ACOX1) deficiency, D-bifunctional protein (DBP) deficiency, and sterol carrier protein X deficiency (SCPx) are the other disorders included under impaired fatty acid β-oxidation (Arora et al; 2020). SCPx deficiency has been described in one adult patient with

Figure 3: Clues to diagnosis of Zellweger spectrum disorders in different age groups with differential

Figure 4: Different phenotypes, age of onset, and clinical features of X-linked adrenoleukodystrophy
dystonia, cerebellar signs, and motor neuropathy (Ferdinandusse et al., 2006).
Disorders with impaired fatty acids α-oxidation
Refsum Disease (RD): RD is caused due to phytanoyl-CoA hydroxylase deficiency which is the first enzyme involved in the α-oxidation of fatty acids. Patients with RD usually present in late childhood. Clinical features of this disorder are represented in Figure-5. The entire spectrum of clinical manifestations is not seen in all cases.
Disorders of the Glyoxylate cycle
Primary Hyperoxaluria type 1: Symptoms of this disorder manifest from infancy to adulthood but majority of them present in childhood or early adolescence. They present with recurrent nephrolithiasis due to deposition of calcium oxalate and nephrocalcinosis. Death in these cases is due to end-stage renal disease and renal failure.
Bile acid synthesis defects
Acyl-CoA oxidase 2 (ACOX2) deficiency, peroxisomal membrane protein 70 (ABCD3) deficiency, and bile acid-CoA: amino acid N-acyltransferase (BAAT) deficiency are included under this category. ACOX2 deficiency and ABCD3 deficiency present in childhood. Both these disorders have similar clinical features with predominant involvement of the liver (Figure 5). BAAT deficiency presents with itching and steatorrhea.
H2O2 Metabolism
Acatalasemia/Hypocatalasemia: It is caused by either complete or partial loss of catalase activity in erythrocytes.This disorder is usually asymptomatic. In rare cases, it may be associated with oral ulcerations or gangrene, or diabetes mellitus.
The age of onset, clinical features, and features that should lead one to suspect the disorders of peroxisomal dysfunction are outlined in Figure 5.
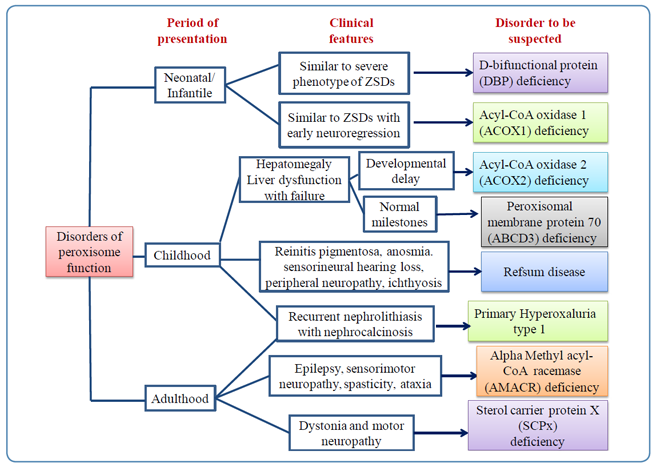
Figure 5: Flow chart showing clues to the diagnosis of disorders of peroxisomal function (single enzyme deficiency disorders)
Diagnosis of peroxisomal disorders
Biochemical workup
For most peroxisomal disorders, biochemical workup involves metabolite assay in plasma and/ or red blood cells(RBCs). Very long-chain fatty acids (VLCFA), docosahexaenoic acid, phytanic acid, and plasmalogen are the biochemical parameters measured by gas chromatography/ mass spectrometry (GCMS). VLCFAs are measured by analyzing the concentration of C26:0, the ratio of C24:0 to C22:0, and the ratio of C26:0 to C22:0. In ZSDs,VLCFA, phytanic acid, pristanic acid,
docosahexaenoic acid, pipecolic acid, and bile acids are elevated in the plasma, and plasmalogens are decreased in the RBCs. An increase in phytanic acid with decreased pipecolic acid in plasma and decreased plasmalogen in RBC is suggestive of RCDP. In Refsum disease, phytanic acid levels are increased but plasmalogen and pipecolic acid levels are normal. In X-ALD, VLCFA is markedly elevated with normal levels of other biochemical substances (Wanders et al) 2018.
Table 3 shows the list of various biochemical substances measured in blood and their levels in different peroxisomal disorders.
Table 3: Biochemical workup for suspected peroxisomal disorders
In plasma | |||||||
Zellweger Spectrum Disorders | RCDP | X-ALD | RD | Fatty acid B-oxidation | |||
Severe | Intermediate | Mild | |||||
VLCFA (C26:0 & C26:1 Ratios of C24, C22 & C26/C22) | Markedly increased | Markedly increased | Increased | Normal | Markedly increased | Normal | Normal except increase in DBP defi- ciency & ACOX1 deficiency |
Phytanic Acid | Markedly increased | Markedly increased | Increased | Markedly increased | Normal | Mark- edly in- creased | Normal except increase in DBP deficiency |
Pristanic acid | Normal to increased | Normal to increased | Normal to increased | Normal | Normal | Normal | Normal ex- cept increase in AMACR & DBP deficiency |
Pipecolic acid | Markedly increased | Markedly increased | Increased | Markedly increased | Normal | Normal | Normal |
Bile acids | Markedly increased | Markedly increased | Increased | Normal | Normal | Normal | Normal except increase in AMACR & DBP deficiency |
In RBC | |||||||
Plasmalogen | Markedly decreased | Markedly de- creased | Decreased | Markedly decreased | Normal | Normal | Normal |
Abbreviations: DBP-D-Bifunctional protein, RCDP-Rhizomelic chondrodysplasia punctata, ALD- Adrenoleukodystrophy, RD-Refsum disease, VLCFA-Very long-chain fatty acids, AMACR-Alphamethyl acyl COA racemase deficiency
X-linked adrenoleukodystrophy is included in neonatal screening programs in several countries (Turk et al., 2020). Recently changes in phospholipid metabolites are found to be reliable biomarkers to indicate neuroinflammation in mice models and X-ALD patients. Further studies need to be done in a large cohort of X-ALD patients to use these metabolites as early biomarkers for neuroinflammation (Kettwig et al., 2021).
In primary hyperoxaluria type 1, the diagnosis is made by the presence of high urinary oxalate excretion and for glycolate oxidase deficiency the diagnosis is by documenting high urinary glycolate levels.
Radiological features
Skeletal abnormalities
Chondrodysplasia punctata at the knee and/ or ankle joints and along the vertebrae in early childhood and rhizomelic shortening are noted in skeletal radiographs in cases with RCDP and Zellweger syndrome (Figure 6). In addition, vertebral cleftsare seen on the skeletal survey in RCDP.
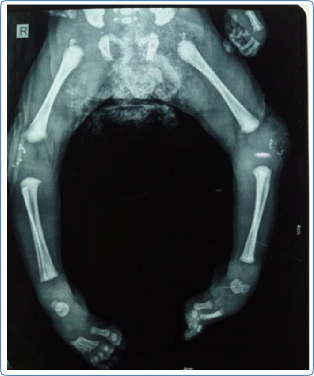
Figure 6: Skeletal radiograph showing chondrody- splasia punctata at the knee joint in a child with rhizomelic chondrodysplasia punctata
Neuroimaging findings
In peroxisomal disorders, specific findings in magnetic resonance imaging (MRI) of the brain are seen only in Zellweger syndrome (Figure 7) and X-ALD. In other peroxisomal disorders, MRI brain findings may provide clues to the diagnosis. MRI brain findings in peroxisomal disorders are listed in Table 4. Typical findings of the MRI brain may not be found in the initial stages of the disease in peroxisomal disorders as they evolve gradually during the disease course. In such a scenario biochemical workup and genetic evaluation would help in the diagnosis.

Figure 7: MRI brain findings in Zellweger syndrome 6A.T2 weighted MRI brain axial view showing germinolytic cysts 6B. T1 weighted MRI brain axial view showing diffuse polymicrogyria
Genetic evaluation
Peroxisomal disorders are rare inherited metabolic disorders. The diagnosis of a specific peroxisomal disorder can be made based on clinical features, biochemical workup, skeletal survey, MRI brain, and molecular genetic testing by whole/ clinical-exome sequencing. Biochemical analysis is required to corroborate the genetic diagnosis in some cases especially in those with variants of uncertain significance.The genomics-first approach
Table 4: MRI brain findings in peroxisomal disorders
Peroxisomal disorder | Findings in MRI Brain | |
Zellweger Spectrum disorders (ZSDs) | Severe phenotype | Cortical migration abnormalities like perisylvian or diffuse polymicrogyria, germinolytic cysts (Figure 6) with or without myelination abnormality |
Intermediate and milder phenotype | Myelination abnormalities; demyelination initially starts in the cerebellum first and later on involves the entire cerebral region | |
Rhizomelic chondrodysplasia punctata (Severe type) | Ventriculomegaly, progressive cerebellar atrophy, delayed myelination of the supratentorial white matter, white matter signal abnormalities in the parieto- occipital region | |
Rhizomelic chondrodysplasia punctata (Milder type) | Normal | |
Cerebral type X-ALD | T2 weighted hyperintensities in parieto-occipital white matter; the frontal, parietal regions are involved rarely | |
Alpha Methylacyl-CoA racemase (AMACR) deficiency | Cerebral atrophy and T2-weighted hyperintensities are noted in the deep white matter of both hemispheres, thalami, midbrain, and pons. | |
Acyl CoA oxidase 1 deficiency and D-bifunctional protein deficiency | Cerebellar atrophy and periventricular white matter hyperintensities (Arora et al., 2020) | |
Sterol carrier protein X deficiency | Bilateral T2 weighted hyperintense signals in the thalamus and pons | |
Refsum disease | No specific MRI findings (Poll-The et al., 2012) | |
would be helpful in cases with milder and atypical phenotypes. A diagnostic flowchart that can be used for peroxisomal disorders is given in Figure 8
.
Management
Surveillance of most of the peroxisomal disorders is by regular monitoring of liver functions, renal and adrenal functions, and annual hearing and ophthalmologic evaluation. In addition, MRI brain is recommended in suspected cases of ZSDs, X-ALD, and also in ACOX1, AMACR, and DBP deficiency.
There is no complete cure available for peroxisomal disorders at present. Supportive therapies like adequate nutrition, physiotherapy, and occupational therapy are to be given. Symptomatic management includes antiepileptic therapy for seizures, and gastrostomy tube feeding for those with feeding difficulty.
Dietary restriction of phytanic acid helps to some extent in patients with Refsum disease. Pyridoxine supplementation has been tried in hyperoxaluria type 1 but has limited success. Good hydration, lithotripsy,andsurgicalinterventionwouldbehelpful to some extent in hyperoxaluriatype 1 but there is a high chance of recurrence of renal stones. In cases with adrenocortical insufficiency, corticosteroid replacement therapy is recommended. In X-linked ALD, Lorenzo’s oil has been tried but has limited success as it does not prevent the progression of neurological symptoms. If done in the early stages, allogeneic hematopoietic stem cell transplantation (HSCT) is shown to either prevent progression or reverse demyelination. Lenti-D gene therapy tried for patients in the early stages of cerebral type of X-ALD showed beneficial results in phase III clinical trial (Eichler et al., 2017). In 2018, Lenti-D™ was heralded as a breakthrough therapy by the United States Food and Drug Administration (US FDA) for treating the cerebral type of X-ALD, as it is found to
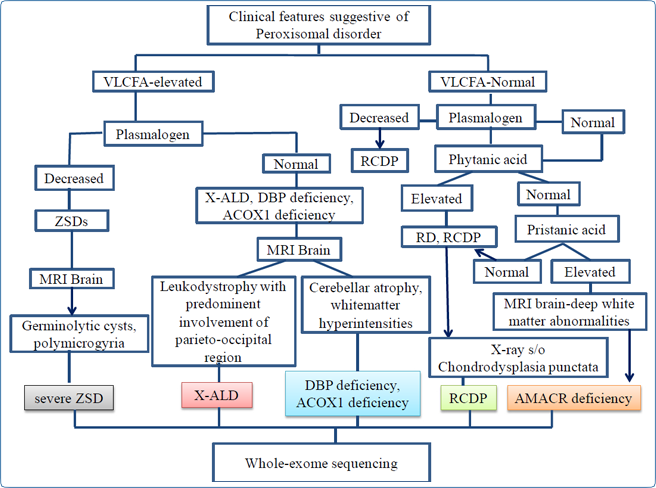
Figure 8: Diagnostic flow chart for peroxisomal disorders
provide significant improvement when compared to other available therapies.
Conclusion
Though most of the peroxisomal disorders can be recognized based on specific clinical clues and diagnostic workup, the milder and atypical phenotypes need a structured clinical and diagnostic approach to establish the exact diagnosis. There is no complete cure for these disorders except for supportive treatment and symptomatic management by a multidisciplinary team. Exact molecular diagnosis, therefore, helps in appropriate genetic counseling and definitive prenatal testing, and also helps the couples to make informed reproductive choices.
References
Arora V, et al. Eyes See what the Mind Knows: Clues to Pattern Recognition in Single Enzyme Deficiency-Related Peroxisomal Disorders. Mol Syndromol. 2020; 11: 309-314.
Eichler F, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017; 377: 1630–1638.
Falkenberg KD, et al. Allelic Expression Imbalance Promoting a Mutant PEX6 Allele Causes Zellweger Spectrum Disorder. Am J Hum Genet. 2017; 101: 965–976.
Ferdinandusse S, et al. Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukoencephalopathy with dystonia and motor neuropathy. Am J Hum Genet. 2006; 78: 1046–1052.
Kettwig M, et al. Targeted metabolomics revealed changes in phospholipids during the development of neuroinflammation in Abcd1(tm1Kds) mice and X-linked adrenoleukodystrophy patients. J Inherit Metab Dis. 2021.44:1174-1185.
Masih S, et al. Twins with PEX7 related intellectual disability and cataract: Highlighting phenotypes of peroxisome biogenesis disorder 9B. Am J Med Genet. 2021; 185: 1504-1508.
Poll-The BT, et al. Clinical diagnosis, biochemical findings and MRI spectrum of peroxisomal disorders. Biochim Biophys Acta. 2012; 1822: 1421-1429.
Takashima S, et al. Expanding the concept of peroxisomal diseases and efficient diagnostic system in Japan. J Hum Genet. 2019; 64:145-152.
Turk BR, et al. X-linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening, and therapies. Int J Dev Neurosci. 2020; 80: 52–72.
Wanders RJA. Peroxisomal disorders: Improved laboratory diagnosis, new defects, and the complicated route to treatment. Mol Cell Probes. 2018;40:60-69.
| Abstract | Download PDF |