| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
GeNeViSTA
| Sl. No. | Age at diagnosis (years) | Sex | Liver (cm palpable) | Spleen (cm palpable) | Hb gm % | Total leucocyte count/ cu mm | Platelet count/ cu mm | Mutation in GBA gene | Plasma chitotriosidase (nmol/ ml/hr) | Beta-glucosidase (normal reference range) | ERT sessions during and after study period | Response to ERT |
| 1 | 8 | F | 6 | 23 | 8.5 | 3200 | 14000 | NA | NA | 1.1 micromol/L of blood/hr (2.3-16) | None | NAP |
| 2 | 2 | M | 0 | 17 | 7.3 | 2300 | 100000 | c.1448T>C; p.L483P | 59922 | 0.608 nmol/hr/mg protein (4-32) | 6 + 11 | CBC: Hb/TLC/ Plt: 9/2.8/1.2; Spleen 12 cm palpable |
| 3 | 1.5 | F | 2 | 8 | NA | NA | NA | NA | NA | 0.8 micromol/L of blood/hr (2.3-16) | None | NAP |
| 4 | 1.5 | F | 5 | 5 | 5.6 | 3000 | 90000 | NA | NA | 0.9 micromol/L of blood/hr (2.3-16) | None | NAP |
| 5 | 3 | M | 9 | 0 | 8.3 | 4000 | 170000 | c.1448T>C; p.L483P | 1041 | 1.9 micromol/L of blood/hr (2.3-16) | 0 + 15 | NAP |
| 6 | 1.1 | M | 2 | 8 | 10.6 | 5500 | 230000 | c.1448T>C; p.L483P | 750 | 0.75 micromol/L of blood/hr (2.3-16) | None | NAP |
| 7 | 2 | M | 5 | 0 | 9.5 | 5000 | 170000 | c.1448T>C; p.L483P | 1361 | 1.58 micromol/L of blood/hr (2.3-16) | None | NAP |
| 8 | 2.5 | M | 0 | 11 | 9.4 | 2750 | 125000 | c.1448T>C; p.L483P | 1178 | 1 micromol/L of blood/hr (2.3-16) | None | NAP |
| 9 | 1.5 | M | 2 | 12 | 8 | 2600 | 90000 | c.1448T>C; p.L483P | 2.79 | 1.8 micromol/L of blood/hr (2.3-16) | None | CBC: Hb/TLC/Plt: 9.5/2.9/1.1; Spleen 8 cm |
| 10 | 3.5 | M | 2 | 10 | 8.3 | 7000 | 190000 | c.1448T>C; p.L483P | 1350 | 0.74 micromol/L of blood/hr (2.3-16) | 0 + 12 | NA; ERT & FU with nearby pediatrician |
| 11 | 1.5 | F | 2 | 7 | 9.3 | 7500 | 210000 | c.1448T>C; p.L483P | 600 | 0.94 micromol/L of blood/hr (2.3-16) | 0 + 12 | NA; ERT & FU with nearby pediatrician |
| 12 | 1.5 | M | 2 | 8 | NA | NA | NA | NA | 12947.3 | 0.43 nmol/hr/mg protein (4-32) | None | NAP |
| 13 | 2 | M | 8 | 12 | 9 | 3750 | 115000 | c.1448T>C; p.L483P | 1232 | 1.38 micromol/L of blood/hr (2.3-16) | None | NAP |
NA - Not available; NAP - Not applicable; M - Male; F - Female; CBC – Complete blood counts; Hb- Hemoglobin; TLC – Total leucocyte count; Plt – Platelet count; ERT – Enzyme replacement therapy; FU – Follow-up
Four patients had history of receiving blood transfusion and two of them had undergone splenectomy before referral. None had history of any bleeding manifestations or any significant bone pain or fracture.
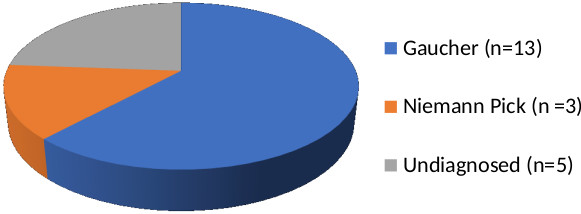
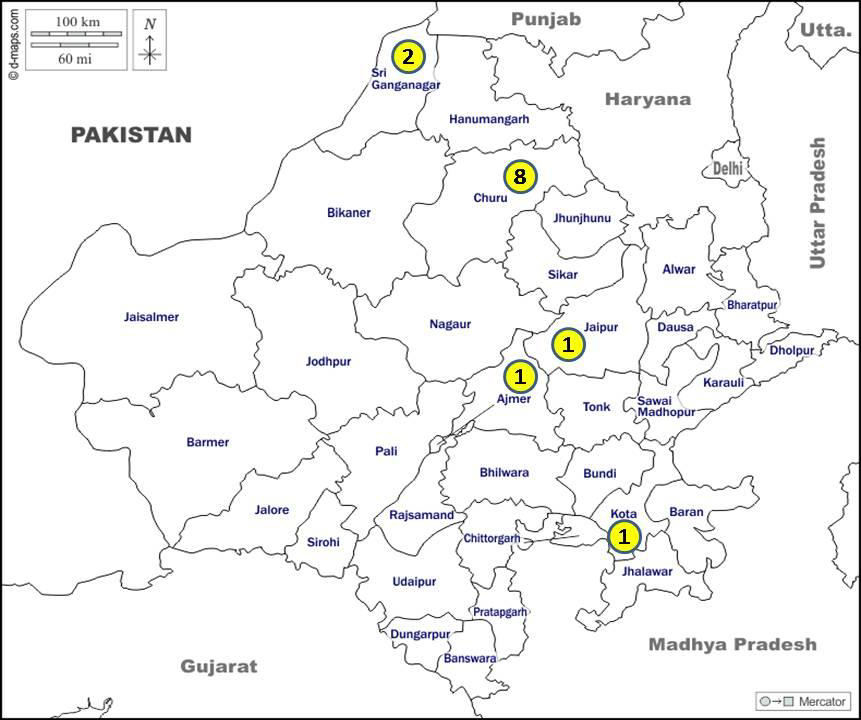
Eight patients of GD hailed from a single district i.e., the Churu district of Rajasthan (Figure 2). Ophthalmological evaluation records were available in 10 children and none of them had cherry red spot or any abnormal eye movements. Hence all our patients were categorized as GD type I. Three children (patient nos. 1, 4 and 9) succumbed to the disease, due to inability to get enzyme replacement therapy (ERT).
There was anemia in all (100 %) with a mean hemoglobin of 8.65 gm/dl and platelets were low (< 1.5 lac/µL) in 4 (30%).
The beta-glucosidase enzyme level in the dried blood spot sample was low in all patients (mean level in patients 1.07 nmol/hr/mL; normal reference range of 2.3-18.4 nmol/hr/mL) suggesting the diagnosis. Out of the 10 patients in whom plasma chitotriosidase assay was done, nine had raised levels and one (patient no. 9) had a normal value, with a mean value of 8931 nmol/ml/hr (range in patients 600 – 59922 nmol/ml/hr; normal reference range of 0-90 nmol/ml/hr).
All nine patients had the same homozygous mutation c.1448 T>C; p.Leu483Pro (previously named as p.Leu444Pro). Though the phenotype of our patients is suggestive of type 1 GD, the genotype i.e., p.Leu483Pro is expected to cause the type III GD phenotype, as per existing data about the mutation. Four patients (patient nos. 2, 5, 10, and 11) are on ERT under the temporary bridging therapy program and have shown improvement in general well-being and spleen size. Complete blood counts were available in two of them and it showed improvement in all parameters. Of the remaining nine patients, three (patient nos. 1, 4, and 9) have succumbed due to inability to get ERT, and the rest are not motivated enough for regular follow up.
GD is the most common lysosomal storage disorder. Type I GD accounts for around 95% of cases. Most published literature is focused on Caucasian patients (Mistry et al., 2015). However, there are a few previous studies available from India. In a large case series of LSD from India, GD was the most common and constituted 16% of the total 387 patients with lysosomal storage diseases reported (Sheth et al., 2013). In a study reported from north India, out of ten cases of GD, eight had type I, and one each had type II and type III (Verma et al., 2012). Another study by Nagral et al. reported type III to be constituting 27% of the total analysed 22 GD patients on ERT (Nagral et al., 2011). Of more than 300 mutations documented in GD, p.Leu444Pro(p.Leu483Pro) appears to be the most prevalent in India (Ankleshwaria et al., 2014).
The metabolic defect in GD is a deficiency of acid beta-glucosidase (also known as lysosomal beta-glucocerebrosidase) enzyme (Cox et al., 1997). The gold standard for diagnosis of GD is acid beta-glucosidase enzyme assay in blood leucocytes. However, dried blood spot (DBS) testing is suggested in India as it overcomes the logistical difficulty in sending blood samples over long distances, in good condition (Verma et al., 2015). Plasma chitotriosidase is commonly employed first as a screening marker for the diagnosis of GD and then as a biomarker for monitoring treatment efficacy. A recent study from India (Kadali et al., 2016) has shown that 22% of our population is deficient in plasma chitotriosidase activity. This could explain the normal chitotriosidase level in one of our patients.
GD is not uncommon in Rajasthan. Geographical clustering of GD in a single district suggests the possible need for population screening in that area. Consanguineous marriage appears to be one predisposing factor. The mutation noted in our series i.e., p.Leu483Pro (p.Leu444Pro) is the most common mutation reported from other studies in India. This mutation is most commonly reported to be associated with the subacute neuronopathic type i.e., type III GD. Thus, though all our patients were phenotypically type I GD, their genotype is consistent with type III GD. It would be interesting to see if some of these patients indeed progress to the type III GD phenotype in the years to come. We plan to do a repeat detailed neurological and ophthalmological evaluation on follow up, to look for subtle abnormalities seen in type III GD.
One of the main limitations of the present study is that being a study from a tertiary referral centre, the results cannot be generalized to the general population.
This article should help improve the awareness of GD when a child presents with splenohepatomegaly of chronic duration when other causes such as infections, portal hypertension and hemolytic anemia have been ruled out.
Acknowledgement: Enzyme replacement therapy for our patients is being provided by Sanofi Genzyme as bridging therapy, on compassionate grounds.
1. Ankleshwaria C, et al. Novel mutations in the glucocerebrosidase gene of Indian patients with Gaucher disease. J Hum Genet. 2014; 59: 223–228.
2. Cox TM, et al. Gaucher’s disease: clinical features and natural history. Baillieres Clin Haematol. 1997; 10(4): 657–689.
3. Kadali S, et al. Clinical evaluation of chitotriosidase enzyme activity in Gaucher and Niemann-Pick A/B diseases: a retrospective study from India. Clin Chim Acta. 2016; 457: 8–11.
4. Mistry PK, et al. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists–oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007; 82(8): 697–701.
5. Mistry PK, et al. Understanding the natural history of Gaucher disease. Am J Hematol. 2015; 90: S6–11.
6. Nagral A, et al. Recombinant macrophage targeted enzyme replacement therapy for Gaucher disease in India. Indian Pediatr. 2011; 48(10): 779.
7. Puri RD, et al. Diagnosis and Management of Gaucher disease in India–Consensus Guidelines of the Gaucher disease Task Force of the Society for Indian Academy of Medical Genetics and the Indian Academy of Pediatrics. Indian Pediatr. 2018; 55(2): 143–153.
8. Sheth J, et al. Burden of lysosomal storage disorders in India: experience of 387 affected children from a single diagnostic facility. JIMD Rep. 2014; 12: 51–63.
9. Verma J, et al. Inherited metabolic disorders: Quality management for laboratory diagnosis. Clin Chim Acta. 2015; 447: 1-7.
10. Verma PK, et al. Spectrum of lysosomal storage disorders at a medical genetics center in northern India. Indian Pediatr. 2012; 49(10):799–804.
| Abstract | Download PDF |