Truncation Variation in the Protocadherin 19 (PCDH19) GeneExhibiting Mosaicism in a Manifesting Heterozygous Male
Truncation Variation in the Protocadherin 19(PCDH19 ) Gene Exhibiting Mosaicism in a Manifesting
Heterozygous Male
Ikrormi Rungsung, Aneek Das Bhowmik, Ashwin Dalal Diagnostics Division, Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India Correspondence to: Dr Ashwin DalalEmail:adalal@cdfd.org.in
1 Abstract
Epilepsy and intellectual disability limited to females (EFMR)/ early infantile epileptic encephalopathy-9 (EIEE9) is an
unusual X-linked disorder in which obligate male carriers are not affected and females show severe epilepsy with cognitive
impairment. In the present study, a male child who presented with cortical dysplasia, gray matter heterotopias and
seizures at two months of age was evaluated using clinical exome sequencing. Clinical exome analysis revealed a mosaic
truncation variant NM_020766.2: c.462 C>G in exon 1 of the protocadherin 19 gene (PCDH19). This
variant was further confirmed by Sanger sequencing, which revealed mosaicism in peripheral blood as well
as saliva DNA in the proband. This variant was not detected in the Sanger sequencing of the parents.
The PCDH19 gene located at the chromosome Xq22.1 locus, encodes for protocadherin delta-2 protein
with 1148 amino acids and is involved in calcium dependent cell-cell adhesion. The present result can be
explained using cellular interference as the disease mechanism and is well supported by previous research
studies.
PCDH19 gene located at the chromosome Xq22.1 locus, spanning six exons and encoding for protocadherin 19
protein (PCDH19), belongs to delta-2 protocadherin subclass of the cadherin superfamily. The PCDH19
gene is predominantly expressed in the brain and several different pathogenic variants have been identified
causing epilepsy and mental retardation limited to females (EFMR). EIEE9 is characterized by febrile
or afebrile tonic-clonic, myoclonic or atonic seizures, starting in the early first year of life, and in some
cases, is associated with intellectual disability. PCDH19 gene is the second known gene related to epilepsy
subsequent to SCN1A gene. The EFMR disorder was first reported in 1971. Later, Dibbens et al identified
PCDH19 as the disease-associated gene in 2008 (Dibbens et al., 2008). EFMR was known to affect only female
carriers and spare hemizygous males. However, affected hemizygous mosaic males were later reported to have
clinical features similar to those in affected females (Depienne et al., 2009). Moreover, a person with sex
chromosome abnormality such as Klinefelter syndrome (47, XXY) or trisomy X syndrome (47, XXX) with
pathogenic variant in PCDH19 gene is also known to develop the phenotype (Romasko et al., 2018). In
addition, there are reports of asymptomatic mosaic males and mutant allele fractions (MAFs) of 4.16%–37.38%
and 1.27%–19.13% in different tissues, hypothesizing 50% of MAFs for disease manifestation (Liu et al.,
2019).
We report on clinical exome sequencing in a four-year-old male child who presented with cortical dysplasia, gray
matter heterotopias and seizures since two months of age which revealed mosaic variant in the PCDH19
gene.
3 Patient and methods
This male patient initially presented at 2 months of age with generalized tonic-clonic seizures without fever. The seizures
were controlled with one drug initially but subsequently the patient needed multiple antiepileptic drugs. Patient had
global developmental delay and at 4 years of age, he could walk without support, run, write a few letters, and could
communicate with the parents. The patient was born to non-consanguineous parents and there was no family history of
seizures. MRI of brain revealed cortical dysplasia, gray matter heterotopias and thickened gray matter. The patient’s
family consented for conducting the study and the same was also approved by the Institutional Ethics
Committee.
Genomic DNA was extracted from the peripheral blood and saliva for the proband and the parents using the
phenol-chloroform and salting out method, respectively. Clinical exome sequencing was performed on the genomic DNA
and sequenced to mean coverage of 100X on the Illumina platform (Centogene, Germany). The reads were mapped
against human reference genome assembly (hg19/GRCh37) using Burrows-Wheeler Aligner (BWA-MEM) and variants
were identified through the Genome Analysis Toolkit (GATK) pipeline. The variants annotated using Annovar were
filtered with 1% minor allele frequency (MAF) against population databases including 1000 genomes, Exome Variant
Server (EVS), Exome Aggregation Consortium (ExAC), Genome Aggregation database (gnomAD), 69 Genome data
(Cg69), Great Middle East (GME_all) and in-house databases. The functional impact of the variants identified was
predicted using bioinformatics tools like PolyPhen2, SIFT and MutationTaster and known mutation databases like
ClinVar, OMIM etc.
Specific genomic primers were designed and PCR was performed on the genomic DNA of proband and parents from
blood and saliva, using the Qiagen Fast Cycling kit (Qiagen, Germany). Sanger sequencing was performed
to confirm the identified variants on the ABI 3130 genetic analyzer machine (Thermo Fisher Scientific,
USA).
4 Results and Discussion
The quality metrics analyses of the clinical exome sequence revealed a mean depth of ˜135X with 97% of the
reads having more than or equal to 20X coverage. The total number of variants was 32,607, which was
reduced to 1333 variants after filtering against population databases. Further, only variants of exonic and
splicing regions were analysed for variant types such as non-synonymous SNV, frameshift deletion, frameshift
insertion, and stop gain variants. A hemizygous variant NM_020766.2(PCDH19):c.462C>G (p.Tyr154*)
was identified in exon 1 of the PCDH19 gene in the proband. This variant is absent in 1000G, ExAC,
gnomAD, Complete genomics (cg69), Great middle east (GME) and in-house Indian databases. At the variant
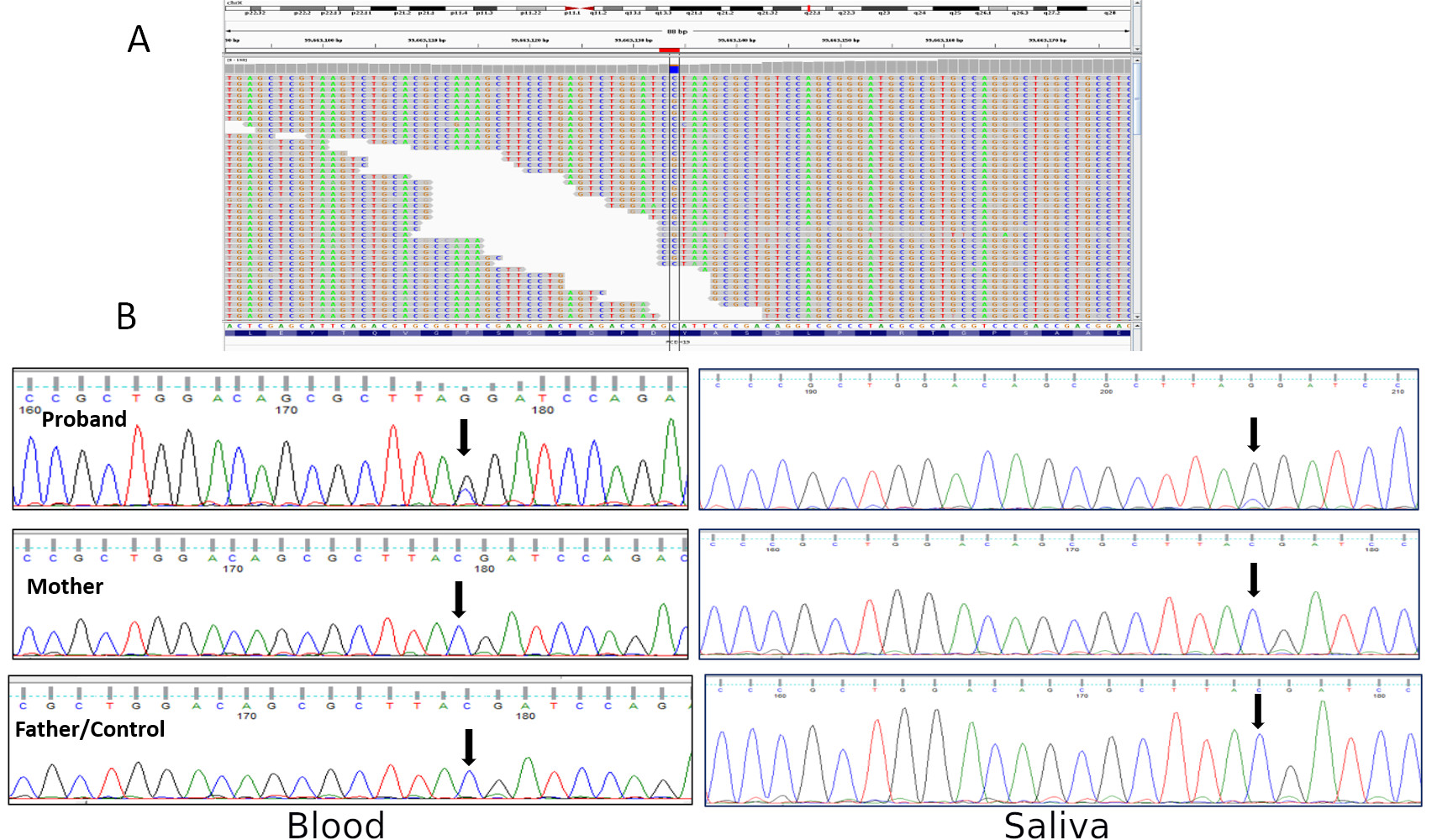
position, the read depths for C and G nucleotides were 16 and 64 respectively (Figure 1A). The identified
variant was confirmed by Sanger sequencing in both the blood and saliva genomic DNA in the proband
(Figure 1B). Parental segregation analysis revealed a de novo mechanism for the variant (Figure 1). It
is also interesting to observe that the mutant G allele showed predominance over the reference C allele
in the targeted Sanger testing. Moreover, the ClinVar database classified the identified variant as likely
pathogenic with the accession ID: VCV000619130.3 from multiple submitters including the submission from
the present study. The other submitters reported on mosaic variants found in five males showing severe
symptoms.
Figure 1: A) Integrative Genomics Viewer (IGV) screenshot showing hemizygous variant c.462C>G in PCDH19
gene in the proband; read count for reference allele C is 16 and alternate allele G is 64. B) Sanger sequencing on
genomic DNA isolated from blood (left panel) and saliva (right panel) showing mosaicism for the variant in the
proband.
The OMIM database has reported PCDH19 gene related phenotype as inherited in an unusual X-linked pattern
(OMIM number. #300088). This unusual mode of inheritance is explained by cellular interference, where the co-existence
of different cellular populations distorts the cell sorting event in male mosaic. The heterozygous females are affected as a
result of random X-inactivation. The cellular interference mechanism was reported from the study of Depienne et al, 2009,
which revealed a deletion in PCDH19 gene in a male patient with similar phenotype of early infantile epileptic
encephalopathy-9 (EIEE9) or ‘epilepsy and mental retardation limited to females’ (EFMR). EFMR has been known to
affect only female patients and pathogenic variation in PCDH19 gene in hemizygous male does not result in the disease.
The PCDH19 gene encodes for the protocadherin-19 protein which is involved in cell-cell adhesion (Juberg et al.,
2009). Hence, cellular interference was hypothesized for the affected females, comprising of two different cell
populations existing as PCDH19-negative and PCDH19-wild type cells due to X inactivation disrupting the cell
sorting event. This cellular interference has also been reported for craniofrontonasal syndrome, caused by
pathogenic variations in the EFBN1 gene. The heterozygous females were observed to be more severely
affected than the hemizygous males. The mosaic males with this condition suffered from a more severe
outcome in X-linked dominant disorder which supports the cellular interference mechanism (Twigg et al.,
2013).
5 Conclusion
Early infantile epileptic encephalopathy-9 (EIEE9) or Epilepsy and mental retardation limited to females (EFMR) is an
epileptic disease characterized by early onset of seizures with or without intellectual disability. The identified variant
NM_020766.2:c.462 C>G in PCDH19 gene in proband is classified as likely pathogenic according to the American College
of Medical Genetics and Genomics/ Association for Molecular Pathology (ACMG/ AMP) guidelines. The aim of this
report is to highlight this neurologic disorder which is inherited through X-linked inheritance with a distinct disease
mechanism known as cellular interference.
References
1. Depienne C, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles
dravet syndrome but mainly affects females. PLoS Genet 2009; 5(2):e1000381.
2. Dibbens LM, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive
impairment. Nat Genet 2008; 40: 776–781.
3. Juberg R. C, et al. A new familial form of convulsive disorder and mental retardation limited to females.
J Pediatr 2009; 79(5), 726–732.
4. Liu A, et al. Mosaicism and incomplete penetrance of PCDH19 mutations. J Med Genet 2019; 56: 81–88.
5. Romasko EJ, et al. PCDH19-related epilepsy in a male with Klinefelter syndrome: Additional evidence
supporting PCDH19 cellular interference disease mechanism. Epilepsy Res 2018; 145: 89–92.
6. Twigg SR, et al. Cellular interference in craniofrontonasal syndrome: males mosaic for mutations in the
X-linked EFNB1 gene are more severely affected than true hemizygotes. Hum Mol Genet 2013; 22: 1654–1662.