Hystrix-like Ichthyosis and Deafness Syndrome in a Toddler
Kanika Singh1, Renu Saxena1, Rishi Parashar2, Sunita Bijarnia-Mahay1* 1Institute of Medical Genetics and Genomics, Sir Ganga Ram Hospital, New Delhi 2Department of Dermatology, Sir Ganga Ram Hospital, New Delhi Correspondence to: Dr Sunita Bijarnia-MahayEmail:bijarnia@gmail.com
1 Abstract
Hystrix-like ichthyosis and deafness (HID) syndrome is characterized by ichthyosis, erythrokeratoderma,
alopecia and deafness in varying degrees of severity. The clinical manifestations are present since birth, evolve
and gradually worsen. It occurs due to a single known mutation in the GJB2 gene. Early diagnosis and
management and genetic counseling require a high index of suspicion for an underlying genetic basis in such skin
disorders.
2 Introduction
Hystrix-like ichthyosis and deafness (HID) syndrome (OMIM#602540) was first described in a patient in 1977 who
presented with icthyosis-hystrix and bilateral hearing loss (Schnyder et al., 1977). Its initial name was ‘ichthyosis hystrix
gravior, type Rheydt’ after the city of origin of the patient, located near Dusseldorf, Germany, with the word ‘hystrix’
indicating spiky porcupine-like skin changes [Konig et al., 1997]. Traupe H suggested including deafness in the
nomenclature, naming it hystrix-like ichthyosis with deafness, or HID syndrome (Traupe, 1989). The molecular basis of
the HID syndrome has been found to be a heterozygous pathogenic variant (p.Asp50Asn) in the GJB2
gene. Pathogenic variants in GJB2 are more commonly known to cause non syndromic deafness [autosomal
recessive (AR) or autosomal dominant (AD)]. A phenotypic variant to the HID syndrome is the keratitis
icthyosis deafness (KID) syndrome. KID syndrome patients have keratitis (inflammation of the cornea) that
can cause photophobia, scarring and vision loss. They also have palmoplantar keratoderma in addition to
erythrokeratoderma, ichthyosis and deafness which is seen in the HID syndrome. About 100 cases of HID have
been reported to date in literature (Avshalumova et al., 2014). Here we present a rare case of the HID
syndrome.
3 Case Report
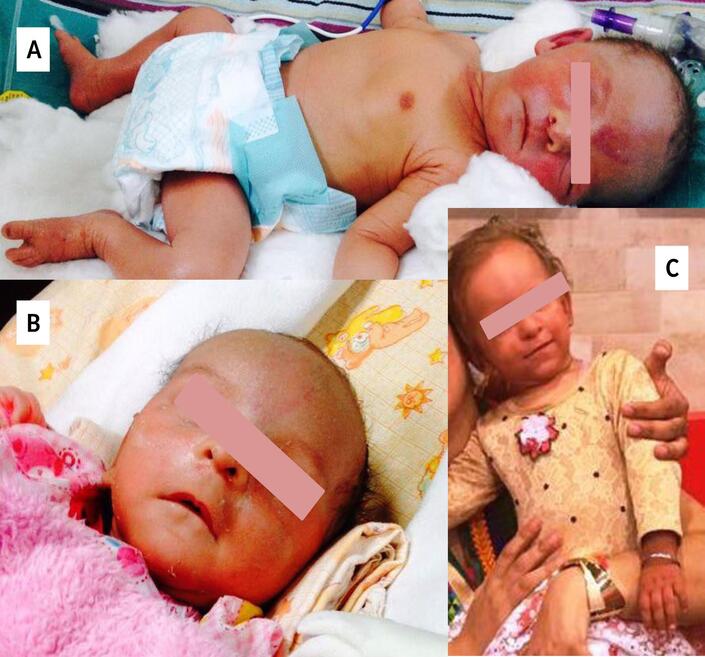
The patient is a 17-month-old girl born to non consanguineous parents. She was born preterm at 36 weeks of gestation,
appropriate for gestation with a birth weight of 2.5 kg. She had required admission in the neonatal intensive care unit
(NICU) for 4 weeks in view of respiratory distress. Soon after birth she developed redness and peeling
of the skin involving the face, arms, trunk and dorsum of hands and feet which persisted at the time of
discharge (Figures 1A and 1B). She was treated for congenital pneumonia and seborrheic dermatitis during her
NICU stay. However, the skin lesions were persistent and difficult to treat. She received multiple courses of
topical steroids, antifungal and antibiotic ointments in view of a possibility of seborrheic dermatitis or atopic
dermatitis along with recurrent skin infections. Over a period of time, she developed diffuse thickening of
the skin and hyperkeratotic plaques over the arms and legs. The eyebrows were absent and hair on the
scalp and body was sparse and lightly pigmented (Figure 1C). There was relative sparing of the skin of the
palms and soles. A lack of sweating was also observed. There was no significant developmental delay. Eye
evaluation did not reveal any significant finding. Her immunoglobulin profile and blood counts were normal. The
patient was the sole affected family member and the only child, with an ongoing pregnancy in the mother.
The family desired a definitive diagnosis for the child and genetic counseling for the ongoing pregnancy.
With a possibility of congenital ichthyotic disorder or ectodermal dysplasia further definitive genetic testing
was planned. Next generation sequencing, for genes related to ichthyosis related disorders revealed the
heterozygous pathogenic variant c.148G>A (p.Asp50Asn/ p.D50N) in the GJB2 gene, which has been
previously reported with HID syndrome. Although the parents did not complain of any significant hearing
impairment in the child, and her speech appeared appropriate for age, a formal hearing test (auditory steady
state responses) showed mild to moderate and moderately profound to severe hearing loss in the right and
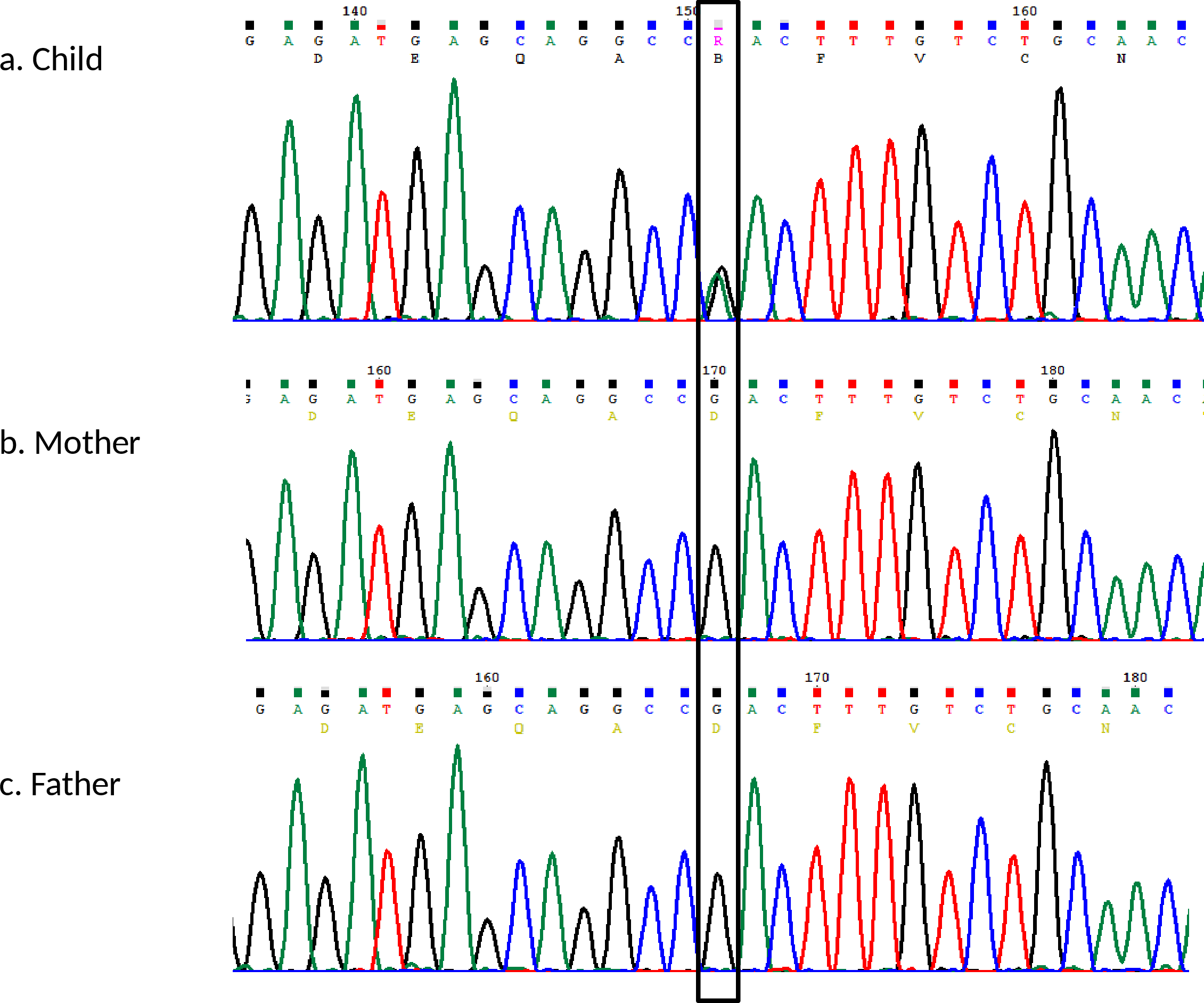
left ear, respectively. Sanger sequencing further confirmed the presence of the mutation in the child. It
was noted to be a de-novo mutation as it was not present in the parents (Figure 2). This confirmed the
overall low risk of recurrence for the ongoing pregnancy (˜1% due to gonadal mosaicism). The parents
chose against prenatal testing of the fetus and continued the pregnancy. For the affected child, the parents
were provided with appropriate dermatological referral and antikeratolytic, antibiotic and emollient topical
treatment. They were advised to discuss the need for hearing aid or cochlear implant in the future, with an
otolaryngologist.
Figure 1: Clinical photographs of the child, in the neonatal period (A, B) showing facial rash and alopecia, and
at 18-months of age (C) showing slight skin rash, and ichthyosis especially on dorsum of hands.
Figure 2: Sanger sequencing analysis of GJB2 gene (A) child heterozygous for the c.148G>A variant; (B & C)
Mother and father negative for the c.148G>A variant.
4 Discussion
HID is a genetic disorder occurring due to a mutation in the GJB2 gene, which belongs to the family of gap junction
proteins. Connexin 26 is a 225- amino acid- long protein encoded by the GJB2 gene located on chromosome 13. Gap
junction channels are made from a family of proteins called connexins. Their main function is to allow passage of small
molecules between adjacent cells, coupling them both metabolically and electrically. The function of the various connexin
channels is distinct in terms of their gating, conductance and permeability characteristics (Avshalumova et al., 2014).
Connexin 26 is involved in intercellular communication and differentiation of cells in the epithelium of
cornea, cochlea, palmoplantar epidermis, hair and sweat glands. The GJB2 gene has been more commonly
implicated with non-syndromic deafness, both autosomal recessive and autosomal dominant types. It has been
identified as the most common cause of non-syndromic deafness – DFNB1 accounting for up to 50% of
congenital severe-to-profound autosomal recessive non-syndromic hearing loss in many countries (Smith et al.,
2016).
GJB2 has also been studied to cause five syndromic forms of deafness that include skin disease. The syndromic
deafness disorders are very rare, and can be divided into two broad groups. The first group includes Bart-Pumphrey
syndrome, Vohwinkel syndrome, and Palmoplantar keratoderma with deafness, presenting with palmoplantar keratoderma
along with deafness. Specifically, patients with the Bart-Pumphrey have nail involvement in the form of leukonychia and
growth on the knuckle pads while, constriction bands and auto amputation have been reported in the Vohwinkel
syndrome (Srinivas et al., 2018).
Hystrix-like ichthyosis deafness syndrome (HID) and keratitis ichthyosis deafness (KID) syndrome make up the second
group. HID manifests shortly after birth with erythematous patches. By the age of 1 year, spiky and cobblestone-like
greyish brown to red hyperkeratotic masses involve the entire skin including the scalp and face. The palms and soles are
usually mildly affected. Scarring alopecia can also occur. Histopathologic features resemble those of lamellar
ichthyosis with reduction of tonofibrils and abundance of mucous granules and are not diagnostic. There
is associated bilateral neurosensory hearing loss. Our patient had all the clinical characteristics of HID
syndrome.
HID and KID are identical at the molecular level and the difference is mainly clinical. Some basic differences between
the two are: KID can present at birth in the form of hyperkeratotic erythroderma which resolves spontaneously only to
recur later but never involves the trunk. Scaling typical of ichthyosis (seen in HID) is not seen, so it is not a true
ichthyosis. In addition, palms and soles are severely affected and eye manifestations are typically seen in KID, although
few case reports have mentioned mild keratitis in patients of HID (Van Geel at al., 2002; Avshalumova et al., 2014). Both
AD (GJB2 gene) and AR (AP1B1 gene) types of KID are known (Boyden et al., 2019). In the AD variety of KID, the
p.Asp50Asn accounts for ˜80% of mutations but other mutations have also been described. KID also has increased
morbidity and chance for disfigurement along with the risk for squamous cell carcinoma (SCC) as compared to HID which
generally has a good prognosis. KID is the only connexin-related skin disorder described with SCC. HID begins as
erythematous patches soon after birth and evolves to ichthyosis involving the scalp and face. Palms and soles are
less affected (differentiating it from KID). A mild punctate keratitis has also been described in some HID
patients. There is only one known mutation for HID (p.Asp50Asn). The electron microscopy features are now
known to not be diagnostic for either disorder, contrary to the previous notion, and include excess formation
of mucous-containing granules and reduction of tonofibrils (Avshalumova et al., 2014). Thus, HID and
KID may represent a spectrum of the same disorder at the molecular level with HID being less severe. As
suspected with other genes with a wide spectrum of disease severity, possible causes include gene-gene
interactions, polymorphisms in other genes expressed in the skin, environmental modifiers and other epigenetic
mechanisms.
A genotype-phenotype correlation has been suggested among the KID patients. The GJB2 p.Asp50Asn
mutation-associated patients of KID syndrome live into adulthood despite vision loss and high risk for developing
squamous cell carcinoma, while the GJB2 p.Gly45Glu and p.Ala88Val mutation-associated patients have higher chances
of dying in childhood due to septic complications (Srinivas et al., 2018).
The other skin disorders reported with connexin mutations are erythrokeratoderma variabilis (EKV), involving
mutations in GJB3 and GJB4, Clouston syndrome (a.k.a. hidrotic ectodermal dysplasia), involving mutations in GJB6,
and oculodentodigital dysplasia (ODDD) caused due to mutations in GJA1 (Avshalumova et al., 2014). Confirmation of
diagnosis in skin disorders has implications for accurate counseling and management. HID syndrome is a condition
that requires regular skin care throughout life. Patients with the KID phenotype need to be monitored for
possibility of developing life-threatening SCC. Accurate diagnosis helped to pick the additional symptom of
hearing loss in our patient, which may have gone unnoticed until significant speech impairment might have
appeared.
Although this is an autosomal dominant disorder, one study reported 64% of cases to be sporadic while 36% cases
were familial, many with unaffected parents (Mazereeuw-Hautier et al., 2007). Hence, germline mosaicism is high for this
disorder, like most skin disorders and this is a challenging point in counselling.
Acknowledgements: The authors thank the parents of the child for their participation, and colleagues for their
guidance.
References
1. Avshalumova L, et al. Overview of skin diseases linked to connexin gene mutations. Int J Dermatol
2014;53:192–205.
2. Boyden LM, et al.Recessive mutations in AP1B1 cause ichthyosis, deafness, and photophobia. Am J Hum
Genet 2019,105:1023–1029.
3. Konig A, et al. Autosomal dominant inheritance of HID syndrome (hystrix-like ichthyosis with deafness).
Europ J Dermatol 1997;7:554–555.
4. Mazereeuw-Hautier J, et al. Keratitis-ichthyosis-deafness syndrome: disease expression and spectrum of
connexin 26 (GJB2) mutations in 14 patients. Br J Dermatol 2007;156:1015–1019.
5. Schnyder UW, et al. Ichthyosis hystrix Typus Rheydt (Fall 19) (Ichthyosis hystrix gravior mit praktischer
Taubheit). Gemeinschafstagung der Sudwestdeutschen Dermatologen-Vereinigung und der Vereinigung
Rheinisch-Westfalischer Dermatologen in Heidelberg vom 8-10, Oktober 1976. Z. Hautkrankheit 1977;
52:762–766.
6. Smith RJH, et al. Nonsyndromic hearing loss and deafness, DFNB1. 1998 Sep 28 [Updated 2016 Aug 18].
In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews[Internet]. Seattle (WA): University of
Washington, Seattle; 1993–2020.
7. Srinivas M, et al. Human diseases associated with connexin mutations. Biochim Biophys Acta
2018;1860:192–201.
8. Traupe H, editor. The Ichthyoses: A Guide to Clinical Diagnosis, Genetic Counseling, and Therapy. Berlin:
Springer-Verlag; 1989: 194–197.
9. Van Geel M, at al. HID and KID syndromes are associated with the same connexin 26 mutation. Br J
Dermatol 2002;146:938–942.