Nail Patella Syndrome: A Case Report and Review of Literature
Vishal Gupta 1, Vinod Kumar 1, Smrithi Salian 1, Hitesh Shah 2and Girisha KM 1* 1Department of Medical Genetics, Kasturba Medical College, Manipal, India 2Department of Orthopedics, Kasturba Medical College, Manipal, India Email:girish.katta@manipal.edu
1 Abstract
Nail patella syndrome is a rare autosomal dominant disorder characterized by nail dysplasia, absent or hypoplastic
patella, abnormality of the elbows and presence of iliac horns. Here, we report a case of a one-and-half-year-old girl with
nail patella syndrome. Mutation analysis of the patient revealed a known de novo pathogenic variant, c.668G>A
(p.R223Q) in exon 4 of the LMX1B gene.
2 Introduction
Nail patella syndrome (NPS) (OMIM 161200) is characterized by absent or hypoplastic, split or ridged and discolored
nails, small and irregular or absent or dislocated patellae, elbow deformity that limits the movements of pronation or
supination and the presence of bilateral iliac horns. Additional features like nephropathy and glaucoma are observed
within the disease spectrum. The prevalence of this condition is about 1 in 50,000 individuals. Haplo-insufficiency of
LMX1B (OMIM 602575) due to the presence of pathogenic variants is known to cause this condition. LMX1B encodes an
LIM homeobox transcription factor 1 beta, which is a member of LIM-homeodomain family. This transcription
factor is essential for the dorsoventral patterning of the limbs, normal development of the kidney, eyes and
dopaminergic and serotonergic neurons. In this article, we report a patient with NPS due to a known variant in
LMX1B.
3 Case report
The patient was a one-and- half-year-old girl. She was the only child born to a non-consanguineous couple at term with no
antenatal or neonatal complications. She had first presented at 19 days of age, with a left club foot deformity and bilateral
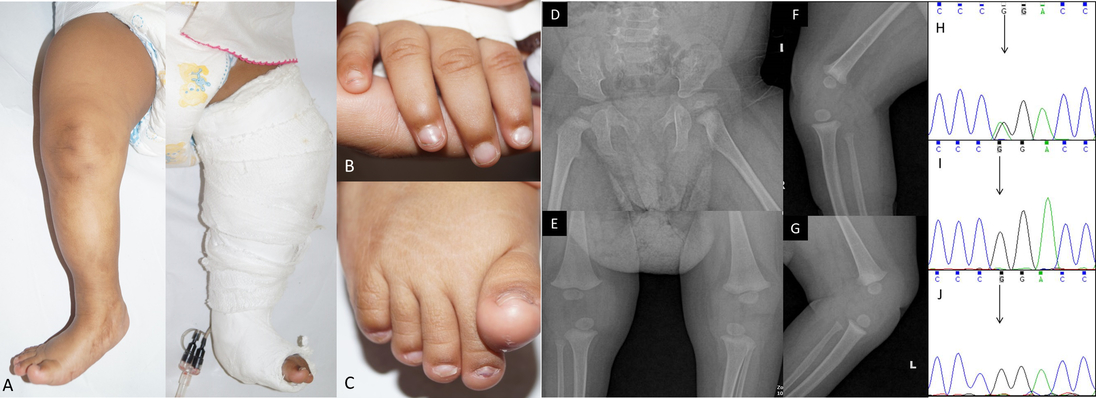
dislocation of knee-caps. On examination she had prominent forehead, depressed nasal bridge, and dystrophic nails with
longitudinal ridges (Fig 1 A-C). Ultrasound examination of both knees showed small patellae with bilateral dislocation.
Radiologic examination of both hips showed bilateral iliac horns (Fig 1 D-G). Rest of the systemic examination was
normal.
Figure 1: Clinical photographs of the patient showing skin dimpling due to the hypoplastic patella and cast
application after tenotomy for congenital talipes equinovarus deformity of the left foot (A), and hypoplastic nails
in the fingers (B) and toes (C). Radiographs showing bilateral iliac horns (D) and anteroposterior (E) and lateral
view of knee joints (F and G). Electropherogram of exon 4 of LMX1B in the patient (H) showing the variant
c.668G>A (indicated by black arrow) and electropherograms of the mother (I) and father (J) showing absence of
this variant (indicating de novo occurrence of the variant).
At two months of age, Achilles tenotomy was done in view of left congenital talipes equinovarus (CTEV). Diagnosis of
nail patella syndrome was made on clinical and radiological grounds. Follow-up at 8 months of age showed normal
developmental milestones and her length was 66cm (normal). Her renal function tests and ophthalmologic evaluations
were within normal limits. The study has the approval of institutional ethics committee and written informed consent was
taken from the patient. We performed Sanger sequencing of LMX1B which revealed a known pathogenic variant
c.668G>A (p.R223Q) in exon 4. Sequence analysis of parents did not reveal this variant suggesting the de novo
occurrence of the mutation (Fig 1 H-J).
4 Discussion
Clinically, the classical presentation of NPS involves changes in nails, knees, elbows and presence of iliac horns. However,
the severity of the disease varies extremely within individuals of the same family also. Many families may remain
undiagnosed because of the mild phenotype. As multiple systems are involved in NPS, there may be a predominance of
disease in one system whereas others may be minimally affected. The spectrum of orthopedic afflictions includes ‘swan
necking’ of index finger, patellar abnormalities (dysplasia, small patellae), tight hamstring muscles and congenital talipes
equinovarus (CTEV). Pinette et al., reported a case of prenatal diagnosis of NPS by identifying skeletal dysplastic changes
(absence of left patella and severe malrotation of left foot) in an anomaly scan establishing the importance of a
targeted anomaly scan in afflicted families. Renal impairment complicates 40% cases of NPS. Primary open
angle glaucoma and ocular hypertension are common ophthalmologic findings. Hence, annual screening for
nephropathy (blood pressure monitoring, urinalysis and urine albumin/ creatinine ratio) and for glaucoma
form part of NPS patient care. In the present study, the patient had three of the classic tetrad of features
typical of NPS. However, there were no signs of elbow deformity, nephropathy, and glaucoma which have
been reported as consistent features of this condition and she would require annual surveillance for the
same.
More than 400 cases with NPS have been reported so far all over the world and more than 140 pathogenic variants
have been identified in patients with NPS. Different types of mutations including missense, nonsense, frameshift, and
splice site mutation, as well as partial and whole gene mutations have been identified in LMX1B. In majority of the cases
(88%) the variants are inherited from a parent in an autosomal dominant manner and in some cases (12%) the mutation
arises de novo. LMX1B regulates expression of genes encoding alpha 3 and alpha 4 chains of collagen IV, interstitial type
III collagen, podocin and CD2AP that form slit pore membrane connecting podocytes. The pathogenic variant identified
in this study is found in the mutation hotspot of LMX1B which spans exons 2-6. Missense mutations are found
to be most commonly present at the mutation hotspot. Therefore, the diagnostic strategy would be to
analyze the mutation hotspot. If no pathogenic variants are found, then deletion/duplication analysis is
considered.
Identification of LMX1B pathogenic variants supports the role of this gene in the causation of NPS and
reinforces the importance of molecular genetic testing as part of prenatal counseling for families with affected
individuals.
References
1. Bongers EM, et al. Nail-patella syndrome. Overview on clinical and molecular findings. Pediatr Nephrol
2002; 17: 703-712.
2. Dunston JA, et al. The human LMX1B gene: transcription unit, promoter, and pathogenic mutations.
Genomics 2004; 84: 565-576.
3. Figueroa-Silva O, et al. Nail-patella syndrome: report of 11 pediatric cases. J Eur Acad Dermatol Venereol.
2016; 30:1614-1617.
5. Lichter PR, et al. Cosegregation of open-angle glaucoma and the nail-patella syndrome. Am J Ophthalmol
1997; 124: 506-515.
6. McIntosh I, et al. Mutation analysis of LMX1B gene in nail-patella syndrome patients. Am J Hum Genet
1998; 63: 1651-1658.
7. Miner JH, et al., Transcriptional induction of slit diaphragm genes by LMX1B is required in podocyte
differentiation. J Clin Invest 2002; 109: 1065-1072.
8. Pinette MG, et al. Early prenatal diagnosis of nail-patella syndrome by ultrasonography. J Ultrasound
Med 1999; 18: 387-389.
9. Sweeney E, et al. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med
Genet 2003; 40: 153-162.
10. Sweeney E, et al. Nail-Patella Syndrome, GeneReviews(R). In: Pagon RA, et al (Eds.), 1993 -. University
of Washington, Seattle. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1132/. Accessed on 10th
March, 2017.