| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
GeNeViSTA
|
|
Cytogenetic abnormalities seen in leukemias are mainly translocations, deletions, ploidy changes and mutations in
specific genes. They differ based on the type of leukemia and are useful in classification and prognosis of the
leukemia.
∙ Acute Myeloid Leukemia: Acute myeloid leukemia (AML) is a tumor of hematopoietic progenitors caused by
acquired oncogenic mutations that impede differentiation, leading to the accumulation of immature myeloid blasts in the
marrow. It is the common form of acute leukemia in adults and accounts for 25% of all leukemias diagnosed in adults, and
is increasingly common with age. Cytogenetic analyses and molecular analyses are currently used to risk-stratify AML.
Cytogenetic abnormalities can be detected in approximately 50% to 60% of newly diagnosed AML (Kumar et al.,
2011).
Genetic abnormalities seen in AML are:
The t(8;21) is more frequent in the young and is rare beyond the age of 50 years. The translocation results in the generation on the derivative chromosome 8 of a consistent hybrid gene, ETOAML- 1 that encodes a novel message for haematopoietic cell proliferation (Rueda et al., 2004).
Most common chromosomal abnormalities t(8;21) and inv(16), disrupt the CBF1α and CBF1β genes, respectively. These two genes encode polypeptides that bind one another to form a CBF1α/CBF1β transcription factor that is required for normal hematopoiesis. The t(8;21) and the inv(16) create chimeric genes encoding fusion proteins that interfere with the function of CBF1α/CBF1β and block the maturation of myeloid cells.
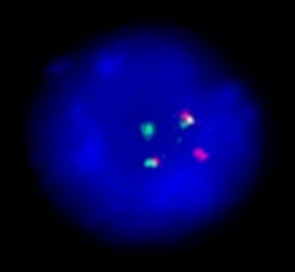
The t(15;17) translocation is found in approximately 95% of (Acute Promyelocytic leukemia) APLs, a specific subtype of AML (Figure 1). The t(15;17) translocation is always associated with APL and leads to the expression of PML-RARα onco-fusion gene in hematopoietic myeloid cells. The PML-RARα onco-fusion protein acts as a transcriptional repressor that interferes with gene expression programs involved in differentiation, apoptosis, and self-renewal. Generally, patients with APL t(15;17) phenotype represent a unique group characterized by distinct biological features and good prognosis, particularly when all-trans retinoic acid (ATRA) is used as part of remission induction (Kumar et al., 2011).
Among the mutated genes FLT3 (ITD) is the most common, seen in one third of AML cases. FLT3 is a receptor
tyrosine kinase that is expressed on hematopoietic progenitor cells and indicative of poor prognosis. Other mutated genes
are NPM1, c-KIT, CEBPA. NPM mutations seen in 30% of cases, it is a protein which normally acts as a
nucleocytoplasmic shuttling protein and regulates the p53 pathway (Verhaak et al., 2005; Stirewalt et al., 2008). NPM1
mutations often coincide with mutations in FLT3, particularly with the ITD-type mutations. The NPM1 mutations in
AML are associated frequently with normal karyotypes. The CEBPA gene provides instructions for making a protein
called CCAAT/enhancer-binding protein alpha. This protein is a transcription factor, which means that it binds to
specific regions of DNA and helps control the activity of certain genes. It is believed to act as a tumor suppressor, helping
to prevent cells from growing and dividing too rapidly or in an uncontrolled way. CEBPA are detected in
15% of patients and associated with favorable response to therapy (Verhaak et al., 2005). Other molecular
markers, such as IDH1, IDH2, and DNMT3A have been suggested to be predictive of risk and response to
treatment.
∙ Acute Lymphoblastic Leukemia (ALL): ALLs are neoplasms composed of the immature T or B cell, called
lymphoblasts. ALL is a malignant clonal proliferation of lymphoid progenitor cells, most commonly of the B-cell
lineage (B-ALL). In the pediatric population, ALL accounts for 81% of childhood leukemias; leukemia overall
accounts for one third of cancers diagnosed in children between ages 0–14 years. Overall, T-ALL has a bad
prognosis when compared to B-ALL. Approximately 90% of ALLs have numerical or structural chromosomal
changes.
Genetic abnormalities seen in B-ALL are:
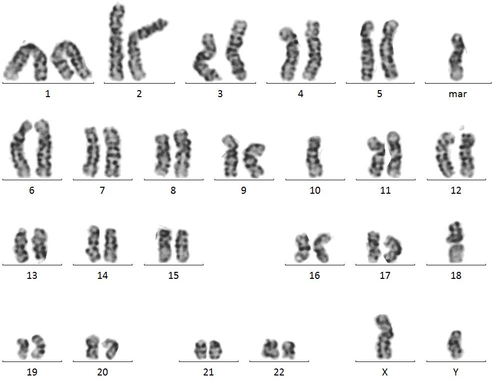
High hyperdiploidy (51–65 chromosomes) is one of the most common cytogenetic abnormalities observed in childhood B-ALL. It is seen in 25-30% of total childhood B-ALL cases, with the highest frequency in the 1 to 4 year age range. High hyperdiploidy is characterized by a nonrandom gain of chromosomes, including +X, +4, +6, +10, +14, +17, +18, and +21. This diagnosis confers a good prognosis in childhood B-ALL (Woo et al., 2014; Ruth et al., 2013). Approximately 20% of hyperdiploid ALL have activating mutations in the receptor tyrosine kinase FLT3. Hypodiploidy is characterized by fewer than 46 chromosomes and is seen in 5-8% of total B-ALL cases (Figure 2). The majority of hypodiploid B-ALL contain 45 chromosomes. The remainder of hypodiploid cases is much rarer and includes high-hypodiploid (40–44 chromosomes).
The t(12;21) is the most common chromosomal rearrangement in ALL. It occurs in 25% of children with B-ALL and confers an excellent prognosis. This translocation joins the TEL (or ETV6) gene on chromosome 12p with the AML1 (or CBFA2) gene on chromosome 21 and is associated with an early pre-B immunophenotype, a younger childhood population, and non hyperdiploidy. The ETV6-RUNX1 fusion protein is thought to disrupt the normal expression of RUNX1-regulated genes by converting RUNX1 to a transcriptional repressor.
The t(1;19)(q23;p13) translocation is found in one fourth of patients with the pre-B immunophenotype. This translocation represents fusion of the E2A and PBX1 genes on chromosomes 1 and 19, respectively. The t(9;22)(q32;q11), or Philadelphia (Ph) chromosome, is present in 20 to 30% of adults and 1 to 5% of children with ALL at the cytogenetic level and is associated with older age, higher leukocyte count, and more frequent CNS involvement at the time of diagnosis (Woo et al., 2014; Ruth et al., 2013).
MLL (mixed-lineage-leukemia) gene rearrangements at 11q23 are present in 80% of all infant B-ALL cases and 10% of all childhood B-ALL. The most common gene rearrangements include t(4;11)(q21;q23) encoding MLL-AF4, t(9;11)(p22;q23) encoding MLL-AF9, t(11;19)(q23;p13.3) encoding MLL-ENL, t(10;11)(p13-14;q14-21) encoding MLL-AF10 and t(6;11)(q27;q23) encoding MLL-AF6 (Woo et al., 2014; Ruth et al., 2013). The t(4;11)(q21;q23) is also associated with high-risk features, most notably a high WBC count and age of onset < 1 year (Hamerschlak et al., 2008).
Conventional karyotyping identifies structural chromosomal aberrations in 50% of T-ALL. Numerical changes are rare, except for tetraploidy which is seen in approximately 5% of cases. The common abnormalities involve the rearrangements involving T-cell receptor genes like deregulation of homeobox genes TLX1 (HOX11) t(10;14)(q24;q11), TLX3 (HOX11L2) t(5;14)(q35;q32), and TAL1 (SCL,TCL5) t(1;14)(p32;q11), t(1;14)(p34;q11) and t(1;7)(p32;q34); deregulation of MYB gene-duplication t(6;7)(q23;q34); and fusion gene rearrangements like PICALM-MLLT10 (CALM-AF10) t(10;11)(p13;q14) and MLL-fusions. Favorable prognosis is associated with subtypes HOX11 or MLL-ENL (Ruth et al., 2013; Hamerschlak et al., 2008).
Up to 70% of T-ALLs have gain-of-function mutations in NOTCH1, a gene that is essential for T-cell development. A
high fraction of B-ALLs have loss-of-function mutations in genes that are required for B-cell such as PAX5, E2A and
EBF.
∙ Chronic Myeloid Leukemia (CML): It is a clonal stem cell disorder characterized by the acquisition of an
oncogenic BCR/ABL fusion protein, usually the result of a reciprocal translocation t(9;22)(q34;q11) and by proliferation
of granulocytic elements at all stages of differentiation. The majority of CML cases are in adults. The frequency of
this type of leukemia is 1 per 1 million children up to the age of 10 years. Among adults, the frequency
is around 1 in 100,000 individuals (Hamerschlak et al., 2008). CML can be divided into three clinically
distinct phases: an initial chronic phase followed by an accelerated phase which subsequently leads to blast
crisis.


Laboratory work up in diagnosis of leukemias includes:
The diagnosis of a hematological neoplasm usually starts from a clinical suspicion, although for chronic leukemias the diagnosis is sometimes an incidental one. A blood count and blood film is an essential first step whenever leukemia, lymphoma or other hematological neoplasm is suspected. Diagnosis is based on peripheral smear and confirmation is done by bone marrow examination. The next step in the diagnostic process depends on the clinical features and the specific condition that is suspected. With advances in immunophenotyping and other techniques, the role of cytochemistry in hematological diagnosis has declined considerably. Further typing of leukemia into myeloid or lymphoid either B cell or T cell, requires the analysis of antigen expression by immunophenotyping.
Cytogenetic and molecular genetic studies are done for molecular classification of leukemia and also to know the bad and good prognostic features, which help in the treatment of the disease. Cytogenetic studies include karyotyping and Fluorescence In-Situ Hybridization (FISH) for detection of the deletions and translocations. Molecular analysis includes PCR, Real Time PCR and the mutational analysis for detection of the fusion transcript and mutation in specific genes. The major applications of each technique are:
Prognostic markers help in predicting the survival of the patients and for planning the appropriate treatment for the disease. The presence of favorable markers in a case means that the survival rate of the patient for that particular disease is more and unfavorable markers have less survival rates. Some of the markers are classified as intermediate markers based on their survival rate, which have less clinical implication when compared to bad and good markers and further evaluation is required (Table-3). Genetic prognostic markers for the chronic leukemias are not well established as in the case of acute leukemias. The major prognostic markers are:
| Type of | Favorable | Unfavorable
|
| Leukemia | | |
| ALL | Hyperdiploidy | Hypodiploidy |
| (>50) t(12;21) | t(9;22) | |
| t(4;11) | ||
| t(1;19) | ||
| AML | t(8;21) | -7/del(7q) |
| t(15;17) | -5/del(5q) | |
| inv(16) | +8 | |
| +9 | ||
| CLL | del 13q | del 17p |
| Trisomy 12 | del 11q | |
| CML | Ph chromosome + | Trisomy 8 |
| del 22q | ||
Leukemias are among the commonest cancers worldwide. Their diagnosis depends mainly on the morphology, cytochemistry and immunophenotyping, but cytogenetic and molecular studies are essential for the molecular classification and predicting the prognosis of the disease. Common cytogenetic studies include karyotyping and FISH for finding the translocations and deletions, and molecular studies include PCR, Real Time PCR and mutation analysis of the relevant genes. Hence, for accurate early diagnosis and treatment, a stepwise evaluation of the case with the suitable diagnostic tests is employed for the favorable outcome.
We would like to thank Dr Ashwani Tandon, Nizam’s Institute of Medical Sciences, Hyderabad for kindly providing the karyotype and FISH images.
1. Abdul-Hamid G. Classification of Acute Leukemia. In: Acute Leukemia -The Scientist’s Perspective and Challenge. Mariastefania Antica (Ed.), 2011. ISBN: 978-953-307-553-2.
2. Deininger MW, et al. The molecular biology of chronic myeloid leukemia. Blood 2000; 96: 3343-3356.
3. Di Bacco A, et al. Molecular abnormalities in chronic myeloid leukemia: deregulation of cell growth and apoptosis. Oncologist 2000; 5: 405-415.
4. Forero RM, et al. Genetics of Acute Lymphoblastic Leukemia, Prof. Margarita Guenova (Ed.), InTech, 2013. DOI: 10.5772/55504.
5. Hamerschlak N. Leukemia: genetics and prognostic factors. J Pediatr (Rio J) 2008; 84: S52-57.
6. Hsiao HH, et al. Additional chromosome abnormalities in chronic myeloid leukemia. Kaohsiung J Med Sci 2011; 27: 49-54.
7. Kumar CC. Genetic abnormalities and challenges in the treatment of acute myeloid leukemia. Genes Cancer 2011; 2: 95-107.
8. Lidiane R, et al. Translocation t(8;21)(q22;q22) in Acute Myeloid Leukaemia. Rev Bras Hematol Hemoter 2004; 26: 66-67.
9. Pokharel M. Leukemia: A Review Article. IJARPB. 2012; 2: 397-407.
10. Puiggros A, et al. Genetic abnormalities in chronic lymphocytic leukemia: where we are and where we go. Biomed Res Int 2014; 435983.
11. Stirewalt DL, et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer 2008; 47: 8-20.
12. Vardiman JW, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114: 937-951.
13. Verhaak RG, et al. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood 2005; 106: 3747-3754.
14. Woo JS, et al. Childhood B-acute lymphoblastic leukemia: a genetic update. Exp Hematol Oncol 2014; 3: 16.
15. Yin CC, et al. Recent advances in the diagnosis and classification of myeloid neoplasms–comments on the 2008 WHO classification. Int J Lab Hematol 2010; 32: 461-476.
| Abstract | Download PDF |