GJC2 Variant Identification in Siblings with Pelizaeus-Merzbacher-Like Disease: Illustrative Report Highlighting the Limitations of Exome Sequencing
T Pragna Lakshmi1Neelam Saini2, Prajnya Ranganath2, Ashwin Dalal1 1 Diagnostics Division, Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India 2 Department of Medical Genetics, Nizam’s Institute of Medical Sciences, Hyderabad, India Correspondence to: Dr Ashwin DalalEmail:ashwindalal@gmail.com
1 Abstract
Whole-exome sequencing (WES) has revolutionized genetic diagnosis and has become a powerful tool for identifying
disease-causing variants. However, WES has limitations such as uneven read coverage, which can result in multiple
low-coverage regions in the exome. Here, we report two affected siblings with a phenotype consistent with
Pelizaeus-Merzbacher-like disease 1 (PMLD1), a rare early-onset autosomal recessive disorder caused by biallelic variants
in the GJC2 gene. WES analysis of one affected child initially revealed only a single heterozygous nonsense variant in
exon 2 of the GJC2 gene. However, on reanalysis with a local de novo assembly approach using the GATK
HaplotypeCaller, a second variant - a frameshift insertion - was subsequently identified. This variant was found to be
present in a low coverage region which is why it was missed in the initial analysis. Analysis of the segregation pattern of
the variants in the affected sibling and parents through targeted Sanger sequencing confirmed that the variants were
present in compound heterozygous form in both the affected siblings. Our study emphasizes the importance
of considering the limitations of exome sequencing, especially in terms of low coverage of certain exonic
regions.
Pelizaeus-Merzbacher-like disease-1 (PMLD1), also referred to as hypomyelinating leukodystrophy type 2 (HLD2), is a
rare autosomal recessive neurological disorder caused by abnormal formation or maintenance of the myelin sheath that
surrounds and protects nerve fibers. This can lead to impaired nerve signal transmission and manifestation of disease
symptoms such as developmental delay, progressive spasticity, motor impairment, ataxia and nystagmus, and findings of
hypomyelination on magnetic resonance imaging (MRI) of the brain (Nahhas et al., 2003). Here, we report a family with
Pelizaeus-Merzbacher-like disease 1 (PMLD1), where initial analysis of whole-exome sequence data identified only one
heterozygous pathogenic variant, but subsequent reanalysis detected the second variant in a low-coverage region of the
gene.
3 Patients and Methods
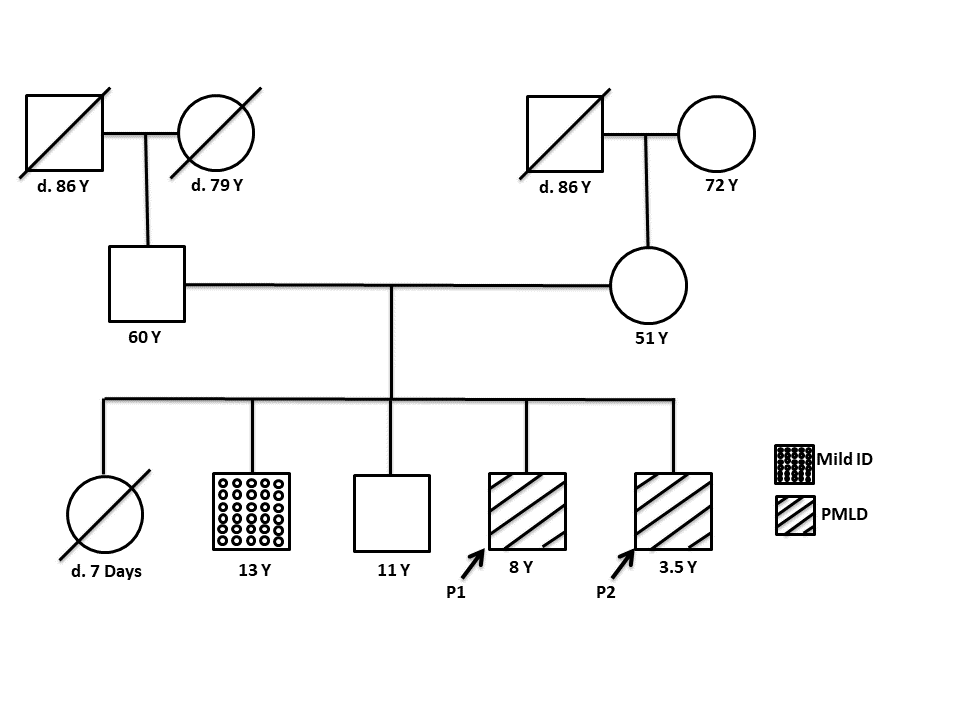
The proband (P1), an 8-year-old male child, was the fourth offspring of non-consanguineous parents (Figure 1). He was
born at term gestation with a birth weight of 2.5 kg and had no history of prenatal or postnatal complications.
During infancy, he had hypotonia and motor developmental delay, with delayed neck holding attained at 11
months and delayed walking without support attained at 2 years. He also had poor vision since infancy. He
had regression of his motor milestones, starting from the age of around 7.5 years. There was no history of
seizures or abnormal movements. The language milestones and intellectual development were normal (IQ-73
at the age of 7 year 7 months). His height was 132 cm (Z score of 0.66), weight was 31 kgs (Z score of
0.87) and head circumference was 49.5 cm (Z score of -2). On examination, he had reduced vision and
nystagmus was present. Increased muscle tone was noted in all four limbs with exaggerated deep tendon reflexes,
positive ankle clonus and bilateral extensor plantar reflex. Other organ systems were normal on clinical
examination. Ophthalmology evaluation revealed optic atrophy with nystagmus. MRI of the brain showed
homogenous T2 hyperintensities involving the central and peripheral aspects of the cerebral white matter as well
as the pons and medulla regions, mild vermian atrophy, and hypoplastic corpus callosum. Based on the
clinical and neuroimaging features, the diagnosis of hypomyelinating leukodystrophy was suspected in the
proband.
Figure 1: The pedigree of the family affected with Pelizaeus- Merzbacher -like disease showing the affected
individuals, the unaffected sibling, and the parents.
His similarly affected 3.5-year-old male sibling (P2) presented with motor developmental delay since infancy along with
nystagmus; his speech was age-appropriate.
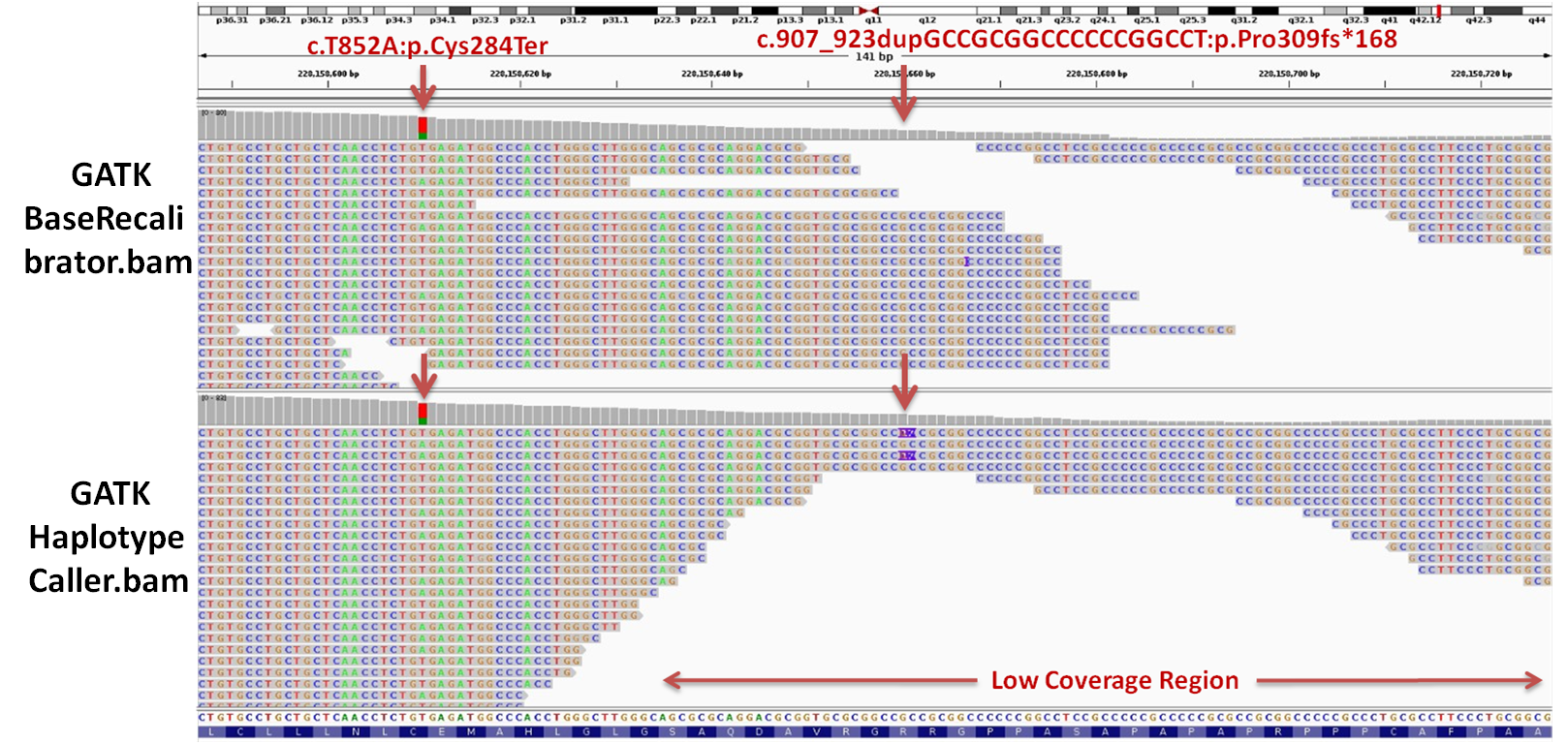
Figure 2: Visualization of bam files generated from the GATK BaseRecalibrator and HaplotypeCaller, using
Integrative Genomics Viewer (IGV).
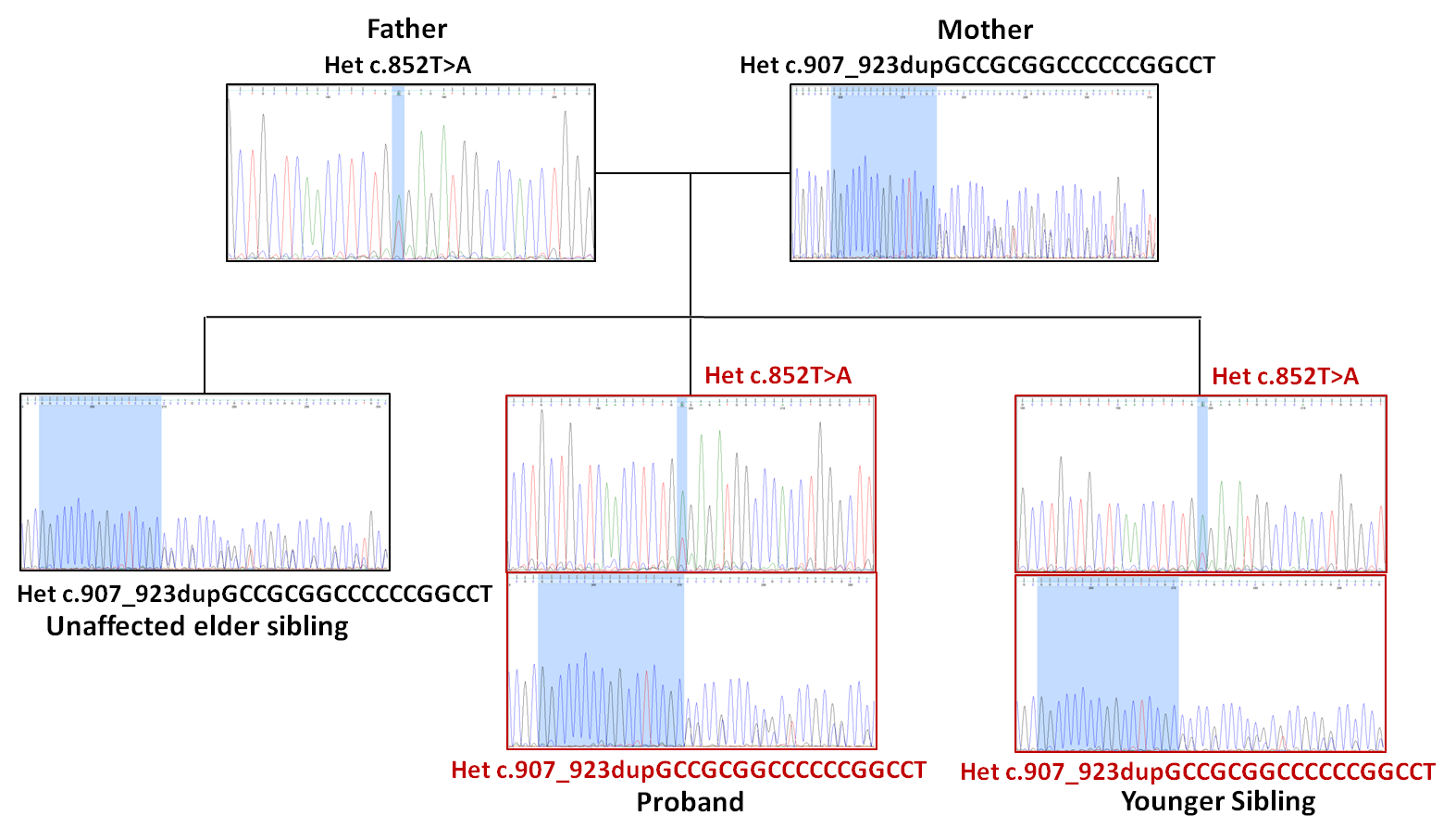
Figure 3: Segregation analysis of the GJC2 compound heterozygous variants within the family, as confirmed by
targeted Sanger sequencing.
WES analysis of the proband P1 initially revealed only a single heterozygous nonsense variant
(GJC2:c.852T>A:p.Cys284Ter) in exon 2 of the GJC2 gene. Subsequent reanalysis of the WES data revealed the second
variant which was a novel frameshift insertion variant (GJC2:c.907_923dup:p.Pro309fsTer168) also in exon 2 of the GJC2
gene. According to the American College of Medical Genetics and Genomics (ACMG) and the Association for
Molecular Pathology (AMP) guidelines, both variants are classified as ‘likely pathogenic’ as they meet the
PVS1 criterion for null variants in a gene where loss-of-function is a known mechanism of disease, and the
c.852T>A variant also meets the PM2 criterion for being absent in large population databases (Richards et al.,
2015). Notably, the frameshift variant had low coverage (depth: 10X) and was not found in the bam file
generated by GATK BaseRecalibrator but was identified in the HaplotypeCaller bam, as depicted in Figure2.
The compound heterozygous status of the affected siblings and the heterozygous carrier status of the unaffected sibling
and both parents were confirmed by targeted Sanger sequencing, supporting an autosomal recessive mode of inheritance
(Figure 3).
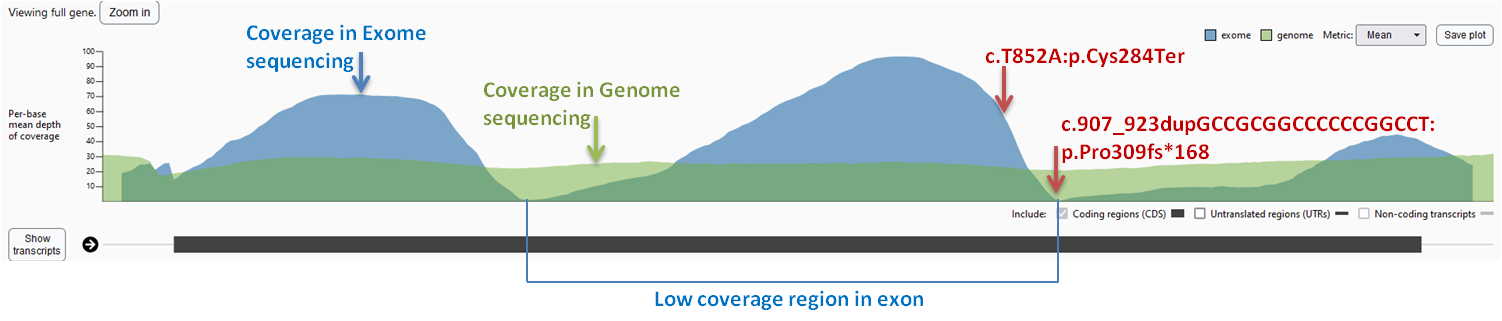
Figure 4: The coverage plot of GJC2 gene in gnomAD v2.1.1. The X-axis represents the genomic position along
the exon2 of GJC2, while the Y-axis represents the depth of coverage. The blue and green regions represent the
coverage obtained from exome and genome sequencing datasets. The variant positions are indicated by arrows.
4 Discussion
The GJC2 gene, which is highly expressed in oligodendrocytes, encodes for the connexin-47 (Cx47) protein that forms gap
junction channels allowing intercellular communication between oligodendrocytes and astrocytes by heterotypic coupling
(Cx47-Cx43 channels) (Kleopa et al.,2004; Qju et al., 2022). Variants in GJC2 can result in loss of function of the
transmembrane protein, which may be attributed to altered channel properties or impaired protein trafficking to the cell
membrane (Biancheri et al.,2013; Owczarek-Lipska et al., 2019). Exon 2 of GJC2, which codes for majority of the Cx47
protein, contains two low-coverage regions in WES data due to its GC-rich sequence. To identify such variants, it is
important to examine the bam file generated by GATK HaplotypeCaller, which uses a local de novo assembly
approach to detect variants, including small insertions and deletions (indels). Nonetheless, the detection
sensitivity of the HaplotypeCaller may vary depending on factors such as sequencing data quality and depth of
coverage.
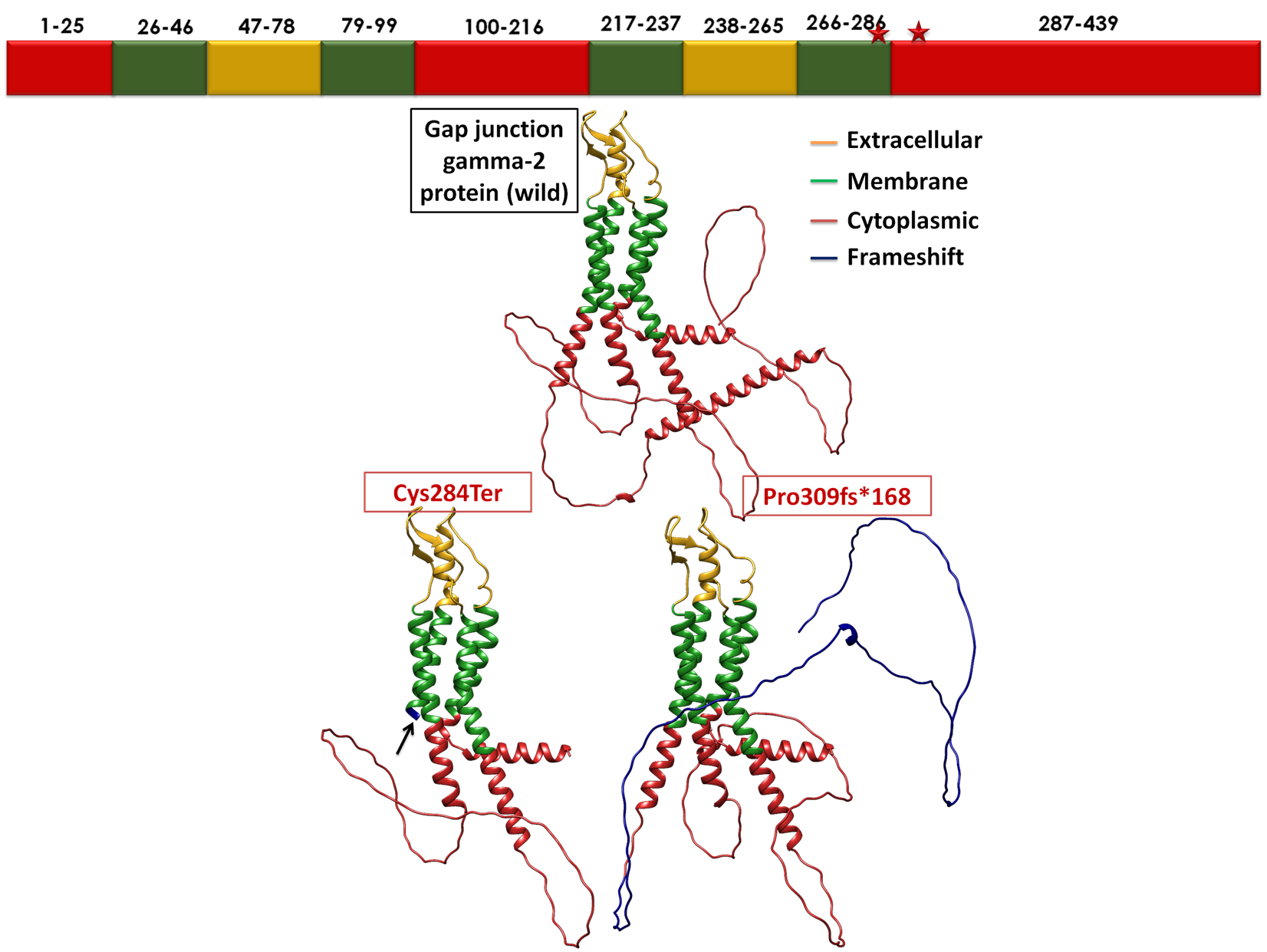
Figure 5: GJC2 protein representation in three forms: wild-type, and in variants p.Cys284Ter and
p.Pro309fsTer168. The extracellular domains are highlighted in mustard yellow, transmembrane domains in green,
and the cytoplasmic domain in red. Both nonsense and frameshift mutations are represented in blue.
A general low coverage of exon 2 region of GJC2 is present in the gnomAD v2.1.1 exome sequencing dataset
(https://gnomad.broadinstitute.org), which may be due to the high GC content of this region (Karczewski et al., 2020).
However, the gnomAD genome sequencing dataset shows uniform coverage in the same region, suggesting that the low
coverage in the exome dataset may be due to limitations of exome sequencing (Figure 4). Variants within such
low-coverage regions can be difficult to call, which makes the uniformly covered exome ‘extracted’ from the
genome sequencing data a helpful approach in such cases. Exome sequence data ‘fetched’ from genome
sequencing data facilitates the identification of such variants that may otherwise be missed in the exome
regions.
The Cx47 protein is composed of two extracellular, four transmembrane and three cytoplasmic domains. The
compound heterozygous variants identified in this family affect the third cytoplasmic domain of Cx47. Specifically, the
nonsense variant (p.Cys284Ter) truncates the protein just before the third cytoplasmic domain, while the frameshift
insertion (p.Pro309fs) is predicted to alter subsequent amino acids and produce an elongated tail of 38 additional residues
in the third cytoplasmic domain (Figure 5). Hence, these variants can have a significant impact on the structure and
function of the Cx47 protein.
5 Conclusion
This report highlights the limitations of exome sequencing in detecting genetic variants in certain exonic regions such as
those with a high GC content and those with repeat elements or segmental duplications. Examining the bam file
generated by GATK HaplotypeCaller would be helpful for identifying variants in such regions. Exome sequence data
extracted from WGS data rather than through WES would be better for such regions due to the uniformity of
coverage.
6 Acknowledgement
The authors thank the family for their participation in the study. This study was supported by the Indian
Council of Medical Research (ICMR)-funded project titled ‘Indian Undiagnosed Diseases Program (I-UDP)’
(33/9/2019-TF/Rare/BMS).
References
1. Biancheri R, et al. Expanded spectrum of Pelizaeus-Merzbacher-like disease: literature revision and
description of a novel GJC2 mutation in an unusually severe form. Eur J Hum Genet. 2013; 21(1): 34-39.
2. Karczewski KJ, et al. The mutational constraint spectrum quantified from variation in 141,456 humans.
Nature. 2020; 581(7809): 434-443.
3. Kleopa KA, et al. Unique distributions of the gap junction proteins connexin29, connexin32, and connexin47
in oligodendrocytes Glia. 2004; 47(4): 346-357.
4. Nahhas N, et al. In: Pelizaeus-Merzbacher-Like Disease 1. 2017 Dec 21 [Updated 2019 Jan 17]. In: Adam
MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington,
Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470716/
5. Owczarek-Lipska M, et al. Novel mutations in the GJC2 gene associated with Pelizaeus-Merzbacher-like
disease Mol Biol Rep. 2019; 46(4): 4507-4516.
6. Qiu Y, et al. Connexin Mutations and Hereditary Diseases Int J Mol Sci. 2022; 23(8): 4255.
7. Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus
recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 2015; 17(5): 405-424.