| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
GeNeViSTA
| Antigenic C1 INH | Functional | C4 | C1q |
||
| Type I HAE | Low | Low | Low | Normal |
|
| Type II HAE | Normal-high | Low | Low | Normal |
|
| HAE III with normal C1-INH | Normal | Normal | Normal | Normal |
|
| Acquired angioedema | Low-normal | Low-normal | Low | Low |
|
| ACE inhibitor-induced angioedema | Normal | Normal | Normal | Normal |
|
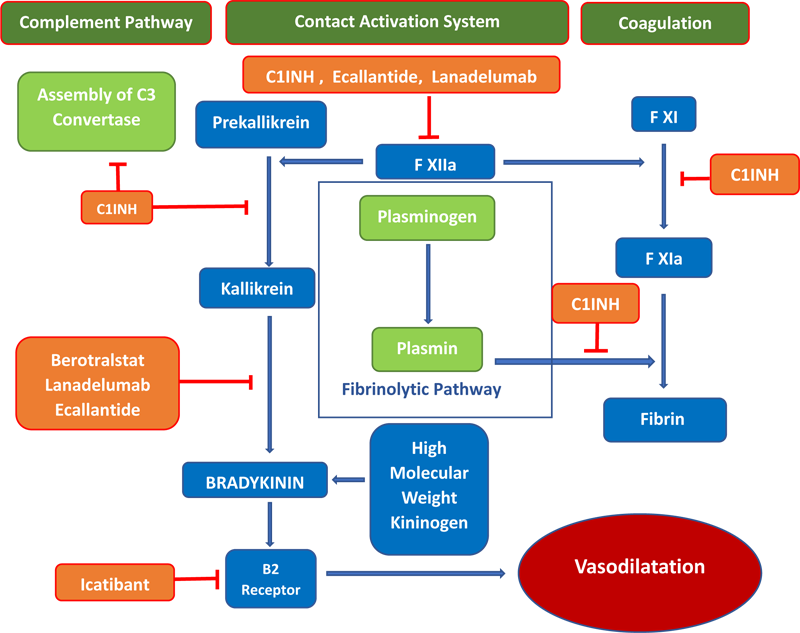
In 1962, Landerman observed a lack of kallikrein inhibitory activity in the plasma of HAE patient plasma. The inherited functional deficiency of C1 INH in affected individuals was first identified by Donaldson and Evans in 1963. Replacement therapy with fresh frozen plasma was tried in 1964-65 as the treatment for acute attacks since the patients did not respond satisfactorily to treatment with epinephrine, antihistaminic agents, or corticosteroids. Subsequently, C1 inhibitors purified from human plasma were found to be effective in both prevention as well as in terminating attacks of angioedema when used as the first specific treatment for HAE in 1973. The use of androgens was the first treatment modality tried for abatement for acute attacks. In 1980, Gadek conducted the first well-controlled study for replacement therapy in 8 HAE patients during an HAE attack by using partly purified C1INH from pooled plasma. In this study, 5 out of 8 patients showed abatement of symptoms in addition to increased serum C4 activity. The first C1 esterase inhibitor therapy (plasma derived C1INH concentrate) to be approved by FDA was Cinryze® in 2008. It is pasteurized with an additional nanofiltration step providing additional protection against enveloped and non-enveloped viral particles and prions. In 2009, Berinert® (a pasteurized plasma derived C1INH concentrate) licensed in Europe for over 20 years, got approval from FDA after the completion of a phase III study for use in the treatment of acute attacks in adolescent and adult patients. Early investigations prepared from the incubation of plasma from HAE patients identified a vascular permeability-enhancing factor named kallikrein assumed to be mediator of swelling in HAE. Trysolol with activity against trypsin, plasmin, and plasma kallikrein was the first plasma kallikrein inhibitor (other than C1INH) to be used. The major drawback associated was severe anaphylactoid reactions. To avoid such adverse effects, a specific plasma kallikrein inhibitor, Ecallantide, FDA-approved in 2009, was used to treat an angioedema attack with lesser risk of anaphylaxis. Inhibition of the binding of bradykinin to its receptor resolves most attacks of acute attack of HAE. Icatibant, FDA-approved in 2011, acts as a specific and selective competitive antagonist of the bradykinin B2 receptor. It can be self-administered as an on-demand treatment of all types of HAE attacks in adults and children with good safety and tolerability. In 2013, Lanadelumab, a fully human monoclonal antibody inhibitor of plasma kallikrein, got FDA global approval in the USA in 2018 after a succession of trials where it was used as a prophylaxis to prevent HAE attacks in patients aged 12 years or more (Syed et al., 2018).
| Approved drug (Generic name) | Indication | Route | Age of patients | Mechanism | Adverse Effects
|
| Plasma-derived C1-INH | On-demand & routine prophylaxis | Intravenous | ≥6 years | Replaces C1-INH | Dysgeusia, injection site reactions, headache, nausea, rash, vomiting, and fever
|
| Recombinant human C1-INH | On-demand & routine prophylaxis | Intravenous | ≥12 years | Replaces C1-INH | Anaphylaxis
|
| Ecallantide | On-demand | Subcutaneous | ≥12 years | Plasma kallikrein inhibitor | Anaphylaxis
|
| Icatibant | On-demand | Subcutaneous | ≥18 years | Bradykinin B2 receptor antagonist | Injection site reaction
|
| Lanadelumab | Routine prophylaxis | Subcutaneous | ≥2 years | Monoclonal antibody (plasma kallikrein inhibitor) | Injection site reactions, upper respiratory infections, headache, rash
|
| Berotralstat | Routine prophylaxis | Oral | ≥12 years | Plasma kallikrein inhibitor | Abdominal pain, vomiting, diarrhoea
|
There are three different principles in the management of HAE. Table 2 summarizes currently available drugs. Due to easy availability and preventive action on plasminogen activation, Tranexamic acid is also used for acute attacks and prophylactically to treat hereditary angioedema. But the data about efficacy is not convincing.
A. On-demand therapy
On-demand therapy is for the treatment of acute attacks with an FDA-approved on-demand HAE medication (Ecallantide, Icatibant, plasma derived C1-INH, or recombinant C1-INH). The drug becomes effective in 60 min with relief in 2 hours. Second dose may be required. In situations of unavailability of specific therapy, solvent-detergent-treated plasma or FFP can be used along with supportive care (intravenous fluids, antiemetics, narcotic pain medication, or intubation).
B. Short-term prophylaxis
Recommended for all medical, surgical, and dental procedures associated with any mechanical impact to the upper aerodigestive tract. Intravenous plasma-derived C1-INH is the first-line short-term prophylaxis drug for HAE with fresh frozen plasma (FFP) as the second-line agent.
C. Long-term prophylaxis
In hereditary angioedema, long-term prophylaxis is indicated for frequent and/or severe episodes of angioedema.
The first attack of HAE occurs in children before 12 years and 23 years of age in 50% and 90% of cases respectively. Most of the attacks are in the form of angioedema of the skin, may manifest as erythema marginatum in 42%–58% of cases. The frequency and severity of attacks may increase during puberty and adolescence. The earlier the onset of symptoms, the more severe the subsequent course of HAE type 1 or 2. Cases may get critical with delayed diagnosis since abdominal attacks may often go unrecognized. Also, asphyxia can ensue rapidly in children, probably because of the small airway diameter. C1-INH, icatibant and ecallantide are approved on-demand treatments for children with HAE 1 or 2.
The symptoms of HAE are more severe in women. There should be cautious use of exogenous steroids in women. During pregnancy, C1-INH is recommended as first-line therapy for pregnant or breastfeeding HAE-1/2 patients. Short term prophylaxis is indicated for procedures. Ecallantide, Lanadelumab and Berotralstat are not recommended in pregnancy. Androgens are contraindicated. Short-term prophylaxis is not indicated in vaginal delivery, since attacks are uncommon during vaginal delivery. But there is increased angioedema of the vulva after delivery. A dose of plasma derived C1-INH is recommended during vacuum or forceps delivery. A preprocedural dose of plasma derived C1-INH is indicated for planned cesarean delivery. General anesthesia should be given with endotracheal intubation.
Plasma derived C1-INH or recombinant C1-INH for on-demand or prophylaxis is recommended. During lactation, anabolic androgens and tranexamic acid are contraindicated with no safety data on the use of Ecallantide, Icatibant, or Lanadelumab.
| IONIS-PKKRx | Antisense inhibitor of prekallikrein and bradykinin production: binds and selectively reduces prekallikrein mRNA in the liver (Ferrone et al, 2019).
|
| NTLA-2002 | Single-dose therapy development; uses in vivo CRISPR-cas9 genome editing. Designed to inactivate the target gene KLKB1 to reduce plasma kallikrein activity.
|
| Garadacimab | Monthly Garadacimab (Fully human, IgG4 monoclonal antibody which targets activated FXII) administration was found to be significantly reducing hereditary angioedema attacks in patients aged 12 years and older (Craiget al., 2023).
|
| KVD824 | Oral small molecule inhibitor of plasma kallikrein. Phase 2 trial was terminated in October 2022 with high level of liver enzymes cited as the reason.
|
| ALN-F12 | Subcutaneously administered Gal-NAc-conjugated siRNA targeting F12 mRNA (ALN-F12) – being done in animal models
|
| PHA-022121 | Small molecule bradykinin 2 receptor antagonist (oral). Results of phase 2 trials indicated that effective bradykinin-inhibiting concentrations can be reached within 15 minutes and maintained for at least 10 hours making it ideally suited for single oral dose treatment of acute HAE attacks (Lesage A et al.,2022).
|
Various therapies to cater to the unmet needs of HAE like long duration of action and oral drugs are in various stages of trial. Table 3 provides an overview of these emerging therapeutic modalities.
Like all other monogenic disorders, cure by gene therapy is the target of research. Ting Qiu et al in 2019, created a heterozygote C1EI deficient mouse model (S63+/-) that shared characteristics associated with HAE in humans including decreased plasma C1EI and C4 levels with increased vascular permeability of skin and internal organs. Single-time intravenous administration of an adeno-associated virus (AAV) gene transfer vector expressing the genetic sequence of the normal human C1 esterase-inhibitor to the gene-deficient mice resulted in sustained human C1EI activity levels above the predicted therapeutic levels and the correction of the vascular leak in the skin and internal organs.
Another promising gene therapy that is being developed for the treatment of HAE with C1-INH deficiency is BMN331. BMN331 is identified as AAV5 hSERPING1, an adeno-associated virus (AAV5)-based gene therapy vector that expresses wild-type human C1 Esterase Inhibitor (hC1-INH), under the control of a liver-selective promoter. BMN331 is currently under a phase 1 / 2 open-label, dose-escalation study to determine the safety tolerability and efficacy.
Currently available therapies are effective and with improved understanding of pathophysiology, newer modalities are in research mode. C1-INH therapy is now available in India and though the cost is high, it has become accessible for Indian patients through funding support provided by the Government of India under the National Policy for Rare Diseases. Awareness about the disease will help in avoiding misdiagnosis. Gene therapy is an exciting approach with promising results in research studies and this is likely to significantly benefit patients with HAE.
1. Agostoni A, et al. Hereditary and acquired C1-inhibitor deficiency: Biological and clinical characteristics in 235 patients. Medicine (Baltimore) 1992; 71: 206-15.
2. Craig TJ, et al. Efficacy and safety of garadacimab, a factor XIIa inhibitor for hereditary angioedema prevention (VANGUARD): a global, multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2023; 401(10382):1079-1090.
3. Ferrone JD, et al. IONIS-PKKRx a Novel Antisense Inhibitor of Prekallikrein and Bradykinin Production. Nucleic Acid Ther. 2019; 29(2): 82-91.
4. Jindal AK, et al. Novel SERPING1 gene mutations and clinical experience of type 1 hereditary angioedema from North India. Pediatr Allergy Immunol. 2021; 32(3): 599-611.
5. Lesage A, et al. In vitro pharmacological profile of PHA-022121, a small molecule bradykinin B2 receptor antagonist in clinical development. Int Immunopharmacol. 2022; 105:108523.
6. Liu J, et al. An investigational RNAi therapeutic targeting Factor XII (ALN-F12) for the treatment of hereditary angioedema. RNA. 2019; 25(2): 255-263.
7. Perumalla S, et al. Clinical profile of hereditary angioedema from a tertiary care centre in India. Indian J Med Microbiol. 2021; 39(4): 509-512.
8. Santacroce R, et al. The Genetics of Hereditary Angioedema: A Review. J Clin Med. 2021; 10(9): 2023.
9. Sinnathamby ES, et al. Hereditary Angioedema: Diagnosis, Clinical Implications, and Pathophysiology. Adv Ther. 2023; 40(3): 814-827.
10. Spaulding W. Methyl Testesterone therapy for hereditary episodic edema (hereditary angioneurotic edema). Ann Intern Med. 1960; 1: 739–744.
11. Syed YY, et al. Lanadelumab: First Global Approval. Drugs. 2018; 78(15):1633-1637.
12. Qiu T, et al. Gene therapy for C1 esterase inhibitor deficiency in a Murine Model of Hereditary angioedema. Allergy. 2019; 74(6):1081-1089.
13. Zhang Y, et al. Exposure-Response Model of Subcutaneous C1-Inhibitor Concentrate to Estimate the Risk of Attacks in Patients with Hereditary Angioedema. CPT Pharmacometrics Syst Pharmacol. 2018; 7(3):158-165.
| Abstract | Download PDF |