Radial Ray Defects: Genetics and Syndromic Etiologies
Sankar VH Genetic Division, Department of Pediatrics, SAT Hospital, Government Medical College, Thiruvananthapuram, Kerala Email:sankarvh@gmail.com
Limb anomalies are a commonly occurring group of malformations, deformations and disruptions due to the
developmental complexity of the limbs, their extended period of morphogenesis and their position outside the body wall.
Limb malformations can be a part of chromosomal aberrations or an array of single gene disorders or may occur due to
environmental teratogens. Radial ray defects are a group of limb malformations characterized by unilateral or
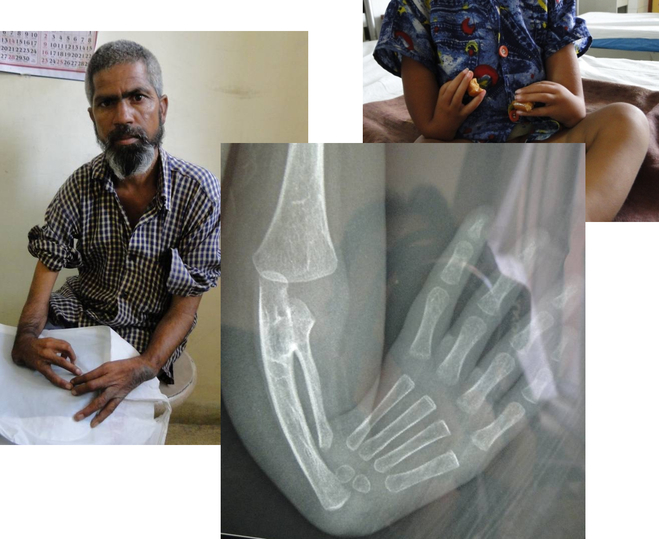
bilateral absence of the radial ray which consists of the radius and thumb (Fig.1). The prevalence of radial
ray defects is low and varies between 1 in 30,000 to 1 in 100,000 with syndromic causes accounting for
approximately two-third of cases.1 The common syndromes associated with radial ray defects are Holt-Oram
syndrome, Fanconi anemia, TAR syndrome and VACTERL association. In addition, chromosomal disorders
such as trisomy 18 can also cause radial ray defects along with significant growth and developmental delay
and other congenital anomalies. In this review syndromes associated with radial ray defects are discussed.
Figure 1: Characteristic hand abnormality in radial ray defects and X-ray showing absent radius and
rudimentary thumb.
1 Molecular Embryology
Limbs develop from embryonic limb buds. Upper limb buds are first visible in the embryo on Day 26-30 as an elevation on
the anterolateral aspects of the body wall. Limb development includes limb initiation and growth (proximo-distal axis)
and its polarization in the antero-posterior and dorso-ventral axis. It involves several coordinated processes characterized
by a constant equilibrium between cell mitotic activity and programmed cell death. Limb bud formation and
growth (proximo-distal axis) are due to rapid cell proliferation in the progress zone (PZ) induced by the
overlying apical ectodermal ridge (AER). The proximo-distal growth is closely linked to polarization along the
antero-posterior axis (under control of the zone of polarizing activity, ZPA) and the dorso-ventral axis (limb
patterning).2
Limb development involves coordinated functioning of various interlinked genes which work by forming a network of
signals. Limb bud outgrowth is promoted by WNT and FGF10. Upper limb anatomy is specified by TBX5 and lower
limb anatomy by TBX4 genes.3 Mutations in T-box genes are associated with syndromes characterized by
limb anomalies, the location of which is in agreement with the expression profile of the respective gene
i.e. in either the arms only (TBX5-Holt Oram syndrome) or both arms and legs (TBX3-ulnar mammary
syndrome). Mutations in the SALL4 gene (SAL-like4), which also encodes a transcription factor, can cause
limb anomalies. Mutations in another gene in the same pathway SALL1 (SAL-like1) are known to cause
Townes-Brocks syndrome. Proximal-distal growth is controlled by the apical ectodermal ridge (AER) whose
formation requires induction by the bone morphogenic protein (BMP) and the homeobox gene MSX2.
The important gene in establishment of antero-posterior polarity is the sonic hedgehog (SHH) gene.4 Its
expression is confined to the ZPA. A number of molecules involved in the SHH pathway are known and
include patched-1, smoothened, GL1-1, GLI-2, GLI-3 and TWIST.5 WNT7A is a major determinant of
dorsal development accomplished through upregulation of LMX1B and WNT7A is repressed by Engrailed 1
(En1).
2 Syndromes with radial ray defects
⋅Holt-Oram Syndrome (OMIM 142900):
Holt Oram syndrome (HOS) is an autosomal dominant disorder
occurring in approximately one in every 10,000 live births and is characterized by cardiac and upper limb malformations.
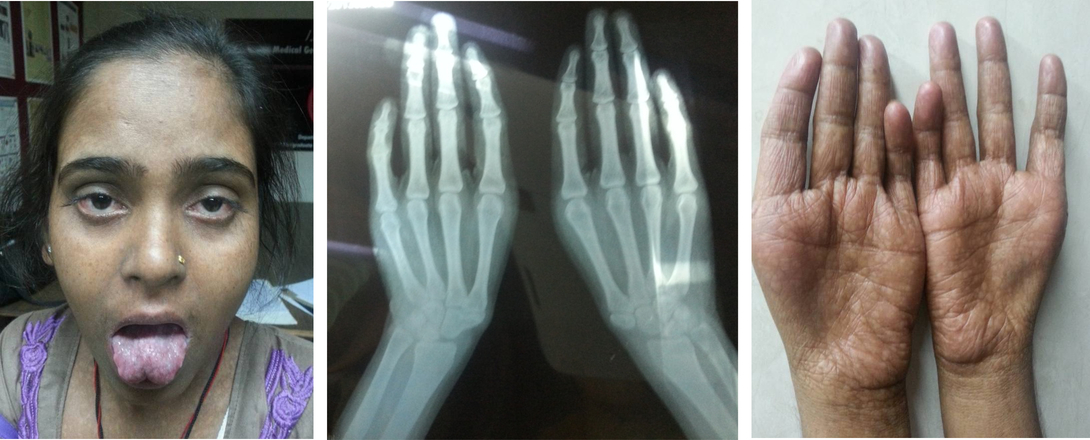
Affected individuals exhibit limb defects that range from subtle carpal abnormalities, absent digits and triphalangeal
thumbs to sloping shoulders and various grades of reduction abnormalities of the radius (Fig.2). Limb defects are usually
bilateral but may be more prominent on the left side. This is frequently associated with cardiac defects like ostium
secundum atrial septal defect, ventricular septal defect or asymptomatic conduction disturbances in most
cases. More complex anomalies like tetralogy of Fallot and pulmonary arterial hypoplasia occur rarely.
Figure 2: Holt Oram Syndrome.
Holt-Oram syndrome is caused by mutations in the TBX5 gene and mutations are spread throughout the gene as
nonsense, insertion, deletion or mis-sense mutations and rearrangements. When applying stringent clinical criteria, a
detection rate of 74% can be achieved in patients with HOS.6 Nevertheless, not all carriers of the TBX5 mutations have
the HOS phenotype, indicating phenotypic heterogeneity at this locus.
⋅Thrombocytopenia with absent radius (TAR) syndrome (OMIM 274000): The thrombocytopenia-absent
radius (TAR) syndrome is a congenital malformation syndrome characterized by bilateral absence of the radii and
thrombocytopenia. Diagnostic criteria by Hall include bilateral absence of the radii in the presence of both
thumbs and a thrombocytopenia. The presence of thumbs distinguishes TAR syndrome from other disorders
featuring radial aplasia, which are usually associated with absent thumbs. Bilateral absence of the radii may be
accompanied by ulnar or humeral anomalies and the most severe cases exhibit phocomelia. Lower limb involvement
is variable (40-47%) and includes dislocation of the patella and/or of the hips, absent tibio-fibular joint,
and lower limb phocomelia. Thrombocytopenia, which may be transient, is seen in all cases and will be
symptomatic in over 90% of cases within the first four months of life. Other systemic problems reported are
cow milk intolerance (60%), renal abnormalities (23%), cardiac abnormalities (15%), genital abnormalities
(3%) and cleft palate. Other associations reported in case series are facial capillary hemangiomas, deafness,
epilepsy and neural tube defects. Differential diagnoses include other conditions with radial ray defects;
however, TAR can be differentiated by the presence of the thumbs in spite of absent radii and other associated
malformation.
TAR syndrome is autosomal recessive in inheritance. An inherited or denovo deletion of 1q21.2 is present in a majority
of cases. However, in view of the apparent autosomal recessive inheritance an additional causative allele should be there
for the development of the disease. A compound inheritance mechanism of a rare null allele and one of two low-frequency
SNPs in the regulatory regions of RBM8A, encoding the Y14 subunit of exon-junction complex (EJC) have been found to
cause TAR. This is the first disease described to be associated with the deficiency of the exon-junction complex
(EJC).7 ⋅Fanconi anemia:
a)
b)
c)
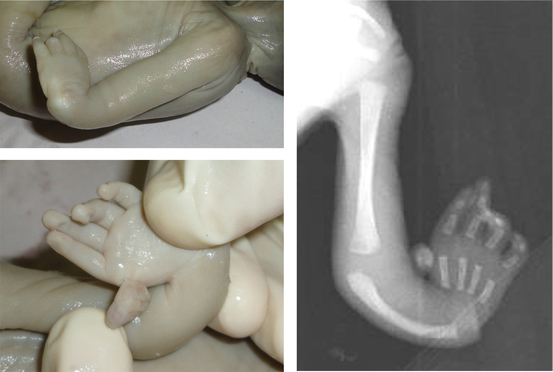
Figure 3: Thumb abnormalities in Fanconi anemia a) triphalangeal thumb b) rudimentary thumb
c) duplication of thumb.
Fanconi anemia (FA) is characterized by physical abnormalities, bone marrow failure and an increased risk of
malignancy. Physical abnormalities are present in 65-70% of cases which include short stature, abnormal skin
pigmentation, malformations of the skeletal system and microcephaly. Upper limb malformations include anomalies of the
thumb (35%) (absent, hypoplastic, bifid, duplicated, rudimentary, triphalangeal, long), radii (7%) (absent or hypoplastic
with abnormal thumbs), hands (5%) (flat thenar eminence, absent first metacarpal, clinodactyly, polydactyly) and ulnae
(1%) (dysplastic, short) (Fig.3). Lower limb anomalies are seen in 5% of cases which include toe syndactyly, club feet and
abnormal toes. Developmental delay can occur in 10% of cases. The diagnosis of FA rests on cytogenetic testing for
increased chromosomal breakages or rearrangements and formation of radial figures in the presence of diepoxybutane
(DEB) or Mitomycin C. Molecular genetic testing is complicated by the genetic heterogeneity with at least 15
genes known to be responsible for the FA complementation groups. Most of these genetic abnormalities are
inherited in an autosomal recessive pattern except mutations in the FANCB gene, which show X-linked
inheritance.8 ⋅SALL 4 related disorders: SALL-4 related disorders include the Duane-radial ray syndrome (DRRS), Okihiro
syndrome and acro-renal-ocular syndrome (AROS), phenotypes previously thought to be distinct entities.9 The
Duane-radial ray syndrome (DRRS) and Okihiro syndrome are characterized by radial ray abnormalities which
include hypoplasia/aplasia of radii, hypoplasia/aplasia of thumb, thenar hypoplasia, triphalangeal thumb,
duplication of thumb (preaxial polydactyly) and Duane anomaly (characterized by uni- or bilateral limitation
of abduction of the eye associated with retraction of the globe and narrowing of the palpebral fissure on
adduction). Acro-renal-ocular syndrome (AROS) is clinically established in individuals with radial ray
malformations, renal abnormalities (renal hypoplasia, mild malrotation, ectopia, horseshoe kidney, vesicoureteric
reflux, bladder diverticula) and ocular abnormalities (ocular coloboma, Duane anomaly). Rarely, SALL4
mutations may cause clinically typical Holt-Oram syndrome. Direct sequencing of the complete SALL4 coding
regions (exons1-4) detects mutation in more than 80% of individuals with DRRS and AROS. Exonic or
whole gene deletions by quantitative real time PCR will detect a further 10-15% cases. Most mutations
are private or have been observed in no more than three independent families. Inheritance is autosomal
dominant with 95% penetrance. The proportion of cases caused by denovo mutations is approximately
40-50%.
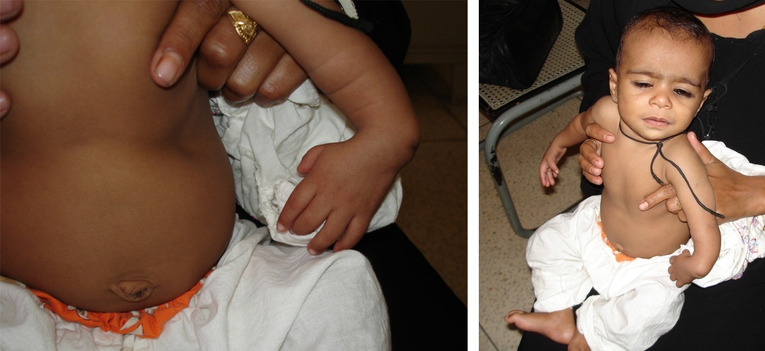
Figure 4: Townes-Brocks syndrome in a father and his son. Hypoplastic radius and absent thumb are seen.
Syndrome
Craniofacialfeatures
Limb anomalies
Other anomalies
Nager acrofacial
dysostosis
(OMIM 154400)
Malar hypoplasia
Micrognathia
Preauricular tag
Cleft palate
Hypoplasia or aplasia of thumb
with or without radius
Proximal radioulnar synostosis
with limitation of elbow
Conductive deafness
Intelligence normal
Rothmund-
Thomson
syndrome
(OMIM 268400)
(Figure 5)
Frontal bossing
Small saddle nose
Prognathism
Small hands and feet
Hypoplastic to absent thumbs
Forearm reduction defects
Mental retardation
Cataract
Sparse hair
Erythema on skin
Poikiloderma
Small dystrophic nails
Baller Gerold
syndrome
(OMIM 218600)
Craniosynostosis
Micrognathia
Microstomia
Epicanthic fold
Hypertelorism
Absent/hypoplsatic radii
Curved ulna
Absent/hypoplastic thumbs
Fused carpal bones
Mental retardation
Congenital heart disease
Renal anomaly
Imperforate anus
RAPADILINO
syndrome
(OMIM 266280)
Long face
Narrow palpebral
fissures
Long slender nose
Cleft palate
Absent thumbs
Joint dislocation
Stiff interphalangeal joints
Small stature
Hearing defect
Infantile diarrhea
Pigmentation
Table 1: Other syndromes with radial ray anomalies.
⋅Townes-Brocks Syndrome (OMIM 107480):
Townes-Brocks syndrome is an autosomal dominant disorder.
Radial ray abnormalities are reported in 50% of published cases. These consist of preaxial polydactyly (bifid thumb),
triphalangeal thumb, hypoplastic thumb, broad thumb, and distal ulnar deviation of the thumb (Fig.4). Anorectal
abnormality is characteristic of this condition. Other abnormalities include auricular, renal and cardiac abnormalities.10
An important differential diagnosis is the VACTERL association, where all these abnormalities can occur.
However, the presence of vertebral defects or tracheo-oeophageal malformation or both would strongly favor
the diagnosis of VACTERL association. Mutation in the SALL1 gene at 16q12.1 is responsible for this
condition.
⋅VACTERL association: VACTERL association comprises Vertebral defects, Anal atresia, Cardiac defects,
Tracheo-Esophageal fistula, Renal malformations, and Limb malformations. There are some single gene
disorders and syndromes which resemble the VACTERL association which include Fiengold syndrome,
22q11 deletion syndrome, Townes-Brocks syndrome and Fanconi anemia. When dysmorphic features, growth
abnormalities and/or learning disability are present, a syndromic diagnosis or chromosomal abnormality has to be
considered.11
Figure 5: Bilateral absent thumb in a case of Rothmund-Thomson syndrome.
3 Testing strategy for individuals with typical radial ray abnormalities
Perform cardiac evaluation, ophthalmologic evaluation and renal ultrasound examination in addition to
routine physical examination.
If no features typical of SALL-4 related disorders are found, molecular genetic testing of the TBX5 gene is
suggested as the first molecular test.
If features typical of SALL-4 related disorders are present, molecular genetic testing of the SALL-4 gene is
suggested as the first step.
If clinical overlap exists with Townes-Brocks syndrome, molecular genetic testing of the SALL1 gene
should be the first test if the radial ray malformations do not include malformations of the radius itself. If
malformation of the radius is present, molecular genetic testing of the SALL4 gene is suggested as the first
molecular test.
4 Prenatal Diagnosis
In pregnancies at risk, detailed high-resolution prenatal ultrasound examination may detect upper-limb malformations
and/or congenital heart malformations. A normal ultrasound examination does not eliminate the possibility of radial ray
defects in the fetus. Prenatal testing for the defect may be most useful in families with a known mutation to confirm
ultrasound findings. If the disease-causing mutation has been identified in the family, prenatal diagnosis for
pregnancies at increased risk is possible by analysis of DNA extracted from fetal cells obtained by amniocentesis
(usually performed at ~15-18 weeks’ gestation) or chorionic villus sampling (usually performed at ~10-12
weeks’ gestation). Because of the significant variable expressivity observed in most conditions especially with
Holt-Oram syndrome both within and among families with the same mutation, the severity of upper-limb
defects and congenital heart malformations cannot be accurately predicted by molecular genetic testing
alone.
5 Acknowledgements
Dr S R Phadke, Department of Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow and Dr
Girisha K M, Department of Genetics, Kasturba Medical College, Manipal for providing photographs for
publication.
References
1. Vergult S, et al. Genet Med 2013; 15: 195-202.
2. Grzeschik KH. Int J Dev Biol 2002; 46: 983-91.
3. Isphording D, et al. Clin Genet 2004; 66: 253-64.
4. Villavicencio EH, et al. Am J Hum Genet 2000; 67: 1047-54.
5. Biesecker LG. J Med Genet 2006; 43: 465-9.
6. McDermott DA, et al. Pediatr Res 2005; 58: 981-6.
7. Albers CA, et al. Curr Opin Genet Dev 2013; 23: 316-23.
8. Zierhut HA, et al. J Genet Couns 2014; 23: 910-21.
9. Kohlhase J. GeneReviews 2004 Aug 16 [updated 2008 Mar 12]. Accessed on 5th Dec 2014.
10. Miller EM, et al. Am J Med Genet A 2012; 158A: 533-40.
 a)
a) 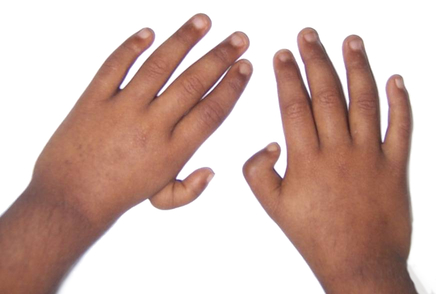 b)
b) 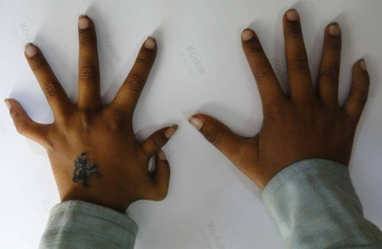 c)
c)