| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
GeNeViSTA
| Gene | Variant details | Zygosity | Splice site tool predictions | OMIM Disease [MIM#] | |
|---|---|---|---|---|---|
| BTD | NM_001370658.1: c.400-3T>G |
Homozygous | Moderate (0.2) | Alteration of the WT acceptor site, most probably affecting splicing | Biotinidase deficiency [253260] |
| CREEBP | NM_004380.3: c.3779+5G>C | Heterozygous | Strong (0.53) | Alteration of the WT donor site, most probably affecting splicing | Rubinstein-Taybi syndrome 1 [180849] |
| SMPD1 | NM_000543.5: c.1341-10_1363dup |
Compound heterozygous with another variant in cis | Strong acceptor gain (0.84) | No significant impact on splicing signals | Niemann-Pick disease, type A [257200] |
| VPS33B | NM_018668.5: c.96G>A p.Gln32= |
Homozygous | Donor loss (0.37) | Alteration of the WT donor site, most probably affecting splicing | Keratoderma-ichthyosis-deafness syndrome, autosomal recessive [620009] |
| MED12 | NM_005120.3: c.887G>A p.Arg296Gln; r.847_888del |
Hemizygous | Acceptor gain (0.38) | Alteration of auxiliary sequences: Significant alteration of ESE / ESS motifs ratio (-9) | Ohdo syndrome, X-linked [300895] |
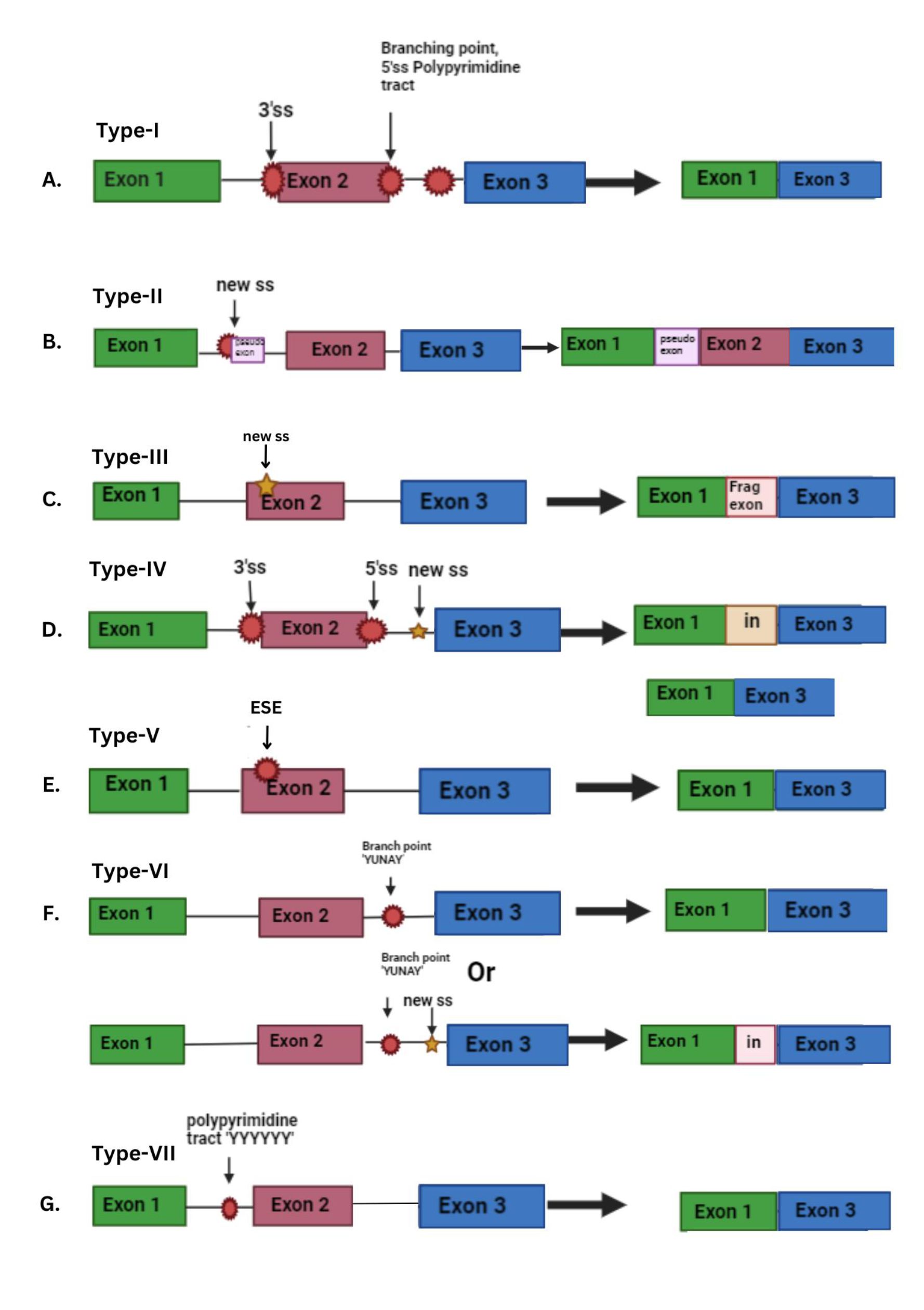
Type 5: Variants that disrupt ESE and lead to skipping of exon
Variants occurring within the exon can also (in contrast to type 3) cause the disruption of exonic splicing enhancers (ESE), which in turn leads to complete exon skipping (Figure 2E). Hence, the presence of exonic changes that result in the interference with exonic splicing enhancers is known as a type V splicing mutation. The utilization of RNA/cDNA sequencing in the diagnosis of genetic diseases is instrumental in identifying type V splicing variants (Ares et al., 1999; Anna et al., 2018). On analysis of NF1 variants in the Leiden Open Variation Database (LOVD), it was found that 69% of the exonic variants were predicted to disrupt ESEs (Anna et al., 2018).
Type 6: Variants at the branch point (YUNAY sequence) results in exon skipping or intron retention
The branch point motif plays a crucial role in the early formation of the spliceosome complex. Changes in the branch point sequence can impact splicing accuracy. The branch point motif, positioned between-9 and - 400 base pairs downstream from the acceptor site, holds significance in humans for early spliceosome complex formation. It carries the consensus sequence YUNAY. Because branch point sequences are inherently variable, variants in this region may result in exon skipping. This occurs due to the improper binding of snRNP splicing proteins, leading to the disruption of the natural acceptor splicing site. Additionally, variants in the branch point sequence can induce intron retention if they generate a new 3’ splice site (Figure 2F). Variants affecting the branch point sequence have been identified in the neurofibromatosis type 1 (NF1) gene. For instance, the variant 2410-18C>G in NF1 leads to the partial retention (17 base pairs) of intron 15. This variant disrupts the original branch point sequence while creating a potential exonic splicing enhancer (ESE). Other splicing variants near this position include 2410-16A>G, 2410-15A>G, and 2410-12T>G. These findings underscore the critical role of this intronic fragment in facilitating the proper splicing of exons 15 and 16 (Anna et al., 2018).
Type 7: Variants in the polypyrimidine tract sequence [(Y)12–17] may lead to splicing alterations
Variants occurring in the polypyrimidine tract (situated upstream of the 3' splice site) or the pyrimidine-rich region (situated downstream of the 5' splice site) can lead to splicing alterations. These types of variants are rare. This sequence is crucial for binding the U2AF65 spliceosome subunit and the polypyrimidine tract-binding protein, both of which play a role in the regulation of alternative splicing (Figure 2G). Variants within the polypyrimidine tract have been observed in Hemophilia B, such as the variant c.253-19_253-16del in F9. This variant leads to the reduction of the polypyrimidine tract length from 24 nucleotides to 20. Consequently, this alteration causes inefficient splicing, resulting in the skipping of exon 3 (Anna et al., 2018).
Point variants at the branch point and polypyrimidine tract are infrequent and challenging to detect when analyzing genomic DNA, especially in coding sequences. Identifying their precise location is difficult, making it challenging to draw conclusions about the potential impact of a specific variant in these regions solely through genomic DNA analysis. To address this, RNA/cDNA sequencing is often employed, or their effects are evaluated through functional studies, such as minigene assays (Anna et al., 2018).
Synonymous variants change the DNA sequence of a gene without affecting the amino acid sequence of the encoded protein. Though these types of variants are considered non-pathogenic, some synonymous variants can affect RNA splicing, translational efficiency, and mRNA stability. Synonymous variants can occur in a gene that has been directly associated with disease pathogenesis, as has been shown in the case of Treacher–Collins syndrome 17, Xlinked infantile spinal muscular atrophy 19, Seckel syndrome 20 and cystic fibrosis (Sauna et al., 2011). One investigation revealed that a synonymous variant within the IL2RG gene resulted in an abnormal splice pattern, leading to decreased expression of the common gamma chain (γc) and the onset of late onset combined immunodeficiency. Another study examined both disease-causing and neutral exonic point variants, concluding that synonymous variants primarily induce disease phenotypes by disrupting splicing. Furthermore, computational predictors were utilized to pinpoint splice-disruptive variants, encompassing missense or synonymous variants. Notably, deep learning-based predictors trained on gene model annotations exhibited the most effective performance in distinguishing disruptive from neutral variants. These findings underscore the significance of considering synonymous variations at splice sites in the investigation of disease genetics (Ares et al.,1999). We identified a synonymous variant, c.96G>A (p. Gln32=) in VPS33B leading to autosomal recessive keratoderma-ichthyosis-deafness (KID) syndrome. The prediction tools were consistent in predicting that this variant could potentially disrupt the WT donor site, and thus be causative (Table 1). It is worth noting here that according to the revised American College of Genetics and Genomics/ Association for Molecular Pathology (ACMG- AMP) recommendations (Walker et al., 2023), synonymous variants which are present in the first or last three bases of the exon and are predicted to impact splicing can be considered as disease-causing, and BP7 criteria should not be applied for the same. This variant in VPS33B mentioned in Table 1 is present in the last base of exon 1, and was thus considered for diagnosis, as the patient had a concordant phenotype.
Bioinformatic approaches
Current methods for detecting and interpreting splice site variants include in-silico tools utilizing machine learning algorithms. Bioinformatics tools such as SpliceFinder integrate functional annotation tools and splice site prediction programs to analyze next-generation sequencing (NGS) data. Position Weight Matrix-based tools are effective in predicting the consequences of variants on mRNA splicing. Recent studies have shown that machine learning classifiers, particularly Random Forest (RF), outperform Support Vector Machine (SVM) in splice site prediction. Additionally, Convolutional Neural Network (CNN) architectures have been developed to predict splice sites and evaluate the impact of genomic variants on splicing. These technologies offer precise and reliable approaches for identifying splice site variants, aiding in the recognition of disease-causing variants and their influence on mRNA splicing (Anna et al., 2018)
These tools were initially created for research purposes but can potentially be integrated into routine diagnostics. They vary in their algorithms, focusing on consensus splicing sites and requiring sequence input within specific positions. Additionally, there are tools designed to assess the impact of distant variants on splicing, predict exon skipping, cryptic site activation, or the generation of aberrant transcripts, as well as algorithms specifically tailored to predict the influence of single nucleotide variants on branch site sequences or polypyrimidine tracts, such as the Branch Site Analyzer and SVM-BP finder (Anna et al., 2018)
When dealing with exonic variants, it is crucial to evaluate their potential effects on exon splicing enhancers (ESEs) or silencers (ESSs). Various algorithms are available for this assessment, such as ESE Finder, and ESRsearch employing a unanimous enrichment approach with hexameric sequence frequencies. Some models like FAS-ESS are based on functional analyses of random sequences through minigene assays, while others like SpliceAid2 rely on the direct interaction between splicing factors and RNA target motifs. Additionally, bioinformatic programs such as mFold or pFold can be employed to predict whether a variant might impact mRNA secondary structure (Chai et al., 2022)
To enhance user convenience, various programs employing different algorithms have been developed and are accessible via websites. Prominent examples include Human Splicing Finder (HSF), Splice AI and SROOGLE, which predict the presence of cis-splicing elements in provided sequences or offer predictions for specific variants in particular genes. Additionally, MutPredSplice is an online tool capable of analyzing individual variants or sets of variants uploaded in a VCF file format. Advanced tools used for annotating variants, particularly those derived from next-generation sequencing data, often integrate splicing prediction algorithms. For instance, the Variant Effect Predictor tool, accessible online, incorporates specialized plugins for splicing analysis utilizing the MaxEntScan model and the dbscSNV matrix from the dbNSFP database (Chai et al., 2022). We have enlisted few examples of splice site variants (Table 1) identified in patients from our centre with the scores of analysis from two commonly used and efficient in-silico prediction tools.
Experimental Techniques
Experimental approaches, including polymerase chain reaction (PCR)-based methods, RNA sequencing (RNA-seq), and mass spectrometry, provide complementary strategies for detecting spliceosome variants at the transcriptomic and proteomic levels. PCR-based assays, such as allele-specific PCR, enable the targeted amplification and quantification of splicing isoforms harboring specific variants. These assays offer high sensitivity and specificity for detecting spliceosome variants in patient samples, facilitating the identification of disease-associated variants and their correlation with clinical phenotypes (Togi et al., 2024)
RNA-seq, on the other hand, offers a genome-wide perspective on alternative splicing events and allows for the identification of novel spliceosome variants in disease-relevant tissues. By profiling the transcriptome of patient samples, researchers can identify dysregulated splicing events and prioritize candidate genes for further functional characterization. Moreover, RNA-seq enables the detection of fusion transcripts resulting from gene fusions and alternative splicing events, providing insights into the molecular mechanisms driving disease pathogenesis (Togi et al., 2024)
Mass spectrometry-based proteomics facilitates the characterization of spliceosome protein complexes and enables the detection of post-translational modifications associated with spliceosome dysfunction. By profiling the proteome of spliceosome complexes, researchers can identify disease-associated variants and elucidate their functional consequences on spliceosome assembly and activity. Furthermore, mass spectrometry enables the quantification of protein expression levels and the identification of dysregulated splicing factors in disease states, providing insights into the molecular mechanisms underlying spliceosome-mediated splicing regulation (Sauna et al., 2011)
Functional assays
Bioinformatic algorithms serve as valuable tools for evaluating potential effects of identified changes. However, it is important to emphasize that these tests provide predictive outcomes, and the precise impact of the variant must be confirmed through functional studies. Another approach to validate the pathogenic effect of a specific splicing variant is to analyze its segregation with the disease in affected and unaffected family members at the DNA level. Nonetheless, laboratory testing is still necessary to ascertain the exact splicing effect (Walker et al., 2023)
The most straightforward and efficient functional assay to ascertain if the chosen variant impacts splicing involves analyzing RNA extracted from pertinent patient tissue or cell lines derived from patient cells. Sequencing RNA/ cDNA following reverse transcription PCR (RT-PCR) enables confirmation of whether the identified variant affects the mRNA sequence. However, a significant challenge with this method is the potential occurrence of nonsense-mediated decay (NMD), which could obscure the effect of the presumed splicing mutation. To mitigate this limitation, patient cells can be treated with NMD inhibitors like puromycin, which halt RNA degradation (Walker et al., 2023)
If suitable material for functional RNA sequencing is not accessible, an alternative option is a minigene assay, a laboratory technique that acts as an in vitro hybrid system enabling ”exon trapping.” This method proves particularly beneficial for analyzing genes with low expression levels in leukocytes or fibroblasts. In the minigene assay, a fragment of the gene under scrutiny, such as a specific exon along with adjacent intronic sequences with and without variants, is amplified and then inserted into a specialized expression plasmid, facilitating the examination of pre-mRNA splicing. This approach serves to validate whether the potential splicing variant impacts splicing efficiency or triggers the activation of alternative cryptic splicing sites. Additionally, it allows for the investigation of the role of cis-acting elements in splicing regulation (Thanapattheerakul et al., 2020)
Lastly, CRISPR-based genome editing technologies enable the generation of isogenic cell lines carrying precise spliceosome variants, allowing for the elucidation of genotype-phenotype correlations and the identification of therapeutic targets. By introducing specific variants into the endogenous genome, researchers can assess the functional consequences of spliceosome variants on pre-mRNA splicing and gene expression regulation. Moreover, CRISPR-based genome editing enables the development of cellular models for studying disease pathogenesis and evaluating therapeutic interventions, paving the way for personalized medicine approaches (Jian et al., 2014) A comprehensive list of all the approaches is enlisted in Table 2.
| Approaches | Tools/ techniques | Website links
|
| 1.Bioinformatic approaches (In-silico prediction tools)
| Human Splice Finder |
|
| Splice AI | https://spliceailookup.broadinstitute.org/
|
|
| Splice site prediction program | www.fruitfly.org/seq_tools/splice.html
|
|
| SPANER | http://tools.genes.toronto.edu/
|
|
| SpliceAid2 | http://193.206.120.249/splicing_tissue.html
|
|
| NetGene2 | http://www.cbs.dtu.dk/services/NetGene2/
|
|
| MutPredSplice | http://www.mutdb.org/mutpredsplice/submit.htm
|
|
| ESE finder |
|
|
| 2.Experimental Approaches
| Polymerase chain reaction-based method |
|
| RNA sequencing (RNA-seq) | https://doi.org/10.1038/s41598-021-89938-2
|
|
| Mass spectrometry | https://doi.org/10.1371/journal.pone.0265766
|
|
| 3.Functional assays
| Minigene splicing assay | https://doi.org/10.1002/humu.22624
|
| Reverse transcription PCR analysis | https://doi.org/10.1186/s12867-016-0060-1
|
|
| Protein truncation test (PTT) | https://10.1007/978-1-59745-388-2_8
|
|
| CRISPR-based genome editing technologies | https://doi.org/10.1002/gcc.22784
|
The detection of spliceosome variants holds significant clinical implications for disease diagnosis, prognosis and therapeutic intervention. Characterization of spliceosome variant profiles in patient populations can inform personalized treatment strategies and guide the selection of targeted therapies tailored to individual molecular profiles. Furthermore, the integration of spliceosome variant detection into routine clinical practice has the potential to revolutionize precision medicine approaches and improve patient outcomes (Hu et al., 2013)
For example, small molecule modulators of spliceosome function, such as splice-switching oligonucleotides and small molecule splicing modulators, hold promise as therapeutic interventions for diseases characterized by aberrant splicing patterns. By targeting specific spliceosome components or splicing regulatory elements, these compounds can modulate splicing efficiency and restore normal gene expression patterns. Clinical trials evaluating the efficacy of spliceosome modulators in various disease settings are currently underway, with promising results reported in preclinical studies (Hu et al., 2013).
Moreover, the identification of spliceosome variants as prognostic biomarkers in cancer and other diseases has important implications for disease management and treatment planning. Patients harboring specific spliceosome variants may benefit from targeted therapies aimed at correcting splicing defects and restoring normal gene expression patterns. Furthermore, the development of companion diagnostic tests for detecting spliceosome variants could enable the stratification of patient populations and facilitate the selection of appropriate therapeutic interventions (Zhai et al., 2013).
Future research directions in spliceosome variant studies encompass a broad spectrum of interdisciplinary approaches, including the development of novel detection methods, elucidation of disease-specific splicing signatures, and exploration of therapeutic modalities targeting spliceosome dysfunction. Integration of multi-omics data, including genomics, transcriptomics, and proteomics, will facilitate a comprehensive understanding of spliceosome-mediated splicing regulation and its implications for human health and disease. Moreover, collaborative efforts between academia, industry, and regulatory agencies are essential for translating basic research findings into clinical applications and improving patient outcomes (Zhai et al., 2013)
Splice site variants can cause diseases by disrupting the proper recognition of exons and altering mRNA splicing. These variants can result in exon skipping, the formation of new exon/intron boundaries, or the activation of cryptic exons. Synonymous variants can also affect splicing by disrupting consensus sequences. To detect splice site variants, various technologies can be used. Bioinformatic algorithms can be applied to predict the effect of identified changes, but functional studies are necessary to confirm the exact impact of specific variants. In vitro transcription and variant analysis via a hybrid minigene system are commonly used methods for functional studies. These approaches can help in the diagnosis of splice site variants and provide insights into the mechanisms underlying splicing-related diseases. By leveraging cutting-edge technologies and interdisciplinary approaches, researchers can elucidate the molecular mechanisms underlying spliceosome-associated diseases and pave the way for innovative diagnostic and therapeutic strategies. Continued investment in spliceosome variant research holds the promise of transformative advances in precision medicine and personalized healthcare.
1. Anna A, et al. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. 2018; 59(3): 253–268.
2. Ares M Jr, et al. A handful of intron-containing genes produces the lion's share of yeast mRNA. RNA. 1999; 5(9):1138-1139.
3. Baralle D. Splicing in action: assessing disease causing sequence changes. J Med Genet. 2005; 42(10): 737–748.
4. Bashari A, et al. Targeting splicing factors for cancer therapy. RNA. 2023; 29(4): 506–515.
5. Chai JN, et al. A splice site mutation associated with congenital CD59 deficiency. Hematol Rep. 2022; 14(2): 172–178.
6. Hauss O, et al. The protein truncation test in mutation detection and molecular diagnosis. In: In Vitro Transcription and Translation Protocols. Totowa, NJ: Humana Press; 2007. p. 151–164.
7. Hu Y, et al. DiffSplice: the genome-wide detection of differential splicing events with RNA-seq. Nucleic Acids Res. 2013; 41(2): e39–e39.
8. Jian X, et al. In silico tools for splicing defect prediction: a survey from the viewpoint of end users. Genet Med. 2014;16(7): 497–503.
9. Sauna ZE, et al. Understanding the contribution of synonymous mutations to human
disease. Nat Rev Genet. 2011; 12(10): 683–691.
10. Taylor J, et al. Mutations in spliceosome genes and therapeutic opportunities in myeloid malignancies. Genes Chromosomes Cancer. 2019; 58(12): 889–902.
11. Thanapattheerakul T, et al. Predicting the effect of variants on splicing using convolutional neural networks. PeerJ. 2020; 8: e9470.
12. Togi S, et al. Qualitative and quantitative analysis of MED12 c.887G>A causing both missense and splicing variants in X-linked Ohdo syndrome. Am J Med Genet A. 2024: e63628.
13. Walker LC, et al. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am J Hum Genet. 2023; 110(7): 1046-1067.
14. Zhai LH, et al. Proteomic characterization of post-translational modifications in drug discovery. Acta Pharmacol Sin. 2022; 43(12): 3112–3229.
| Abstract | Download PDF |