| E-mail ID : info@iamg.in |
| E-mail ID : info@iamg.in |
Online Submission |
| Click Here For Online Submission |
| Instructions for authors |
Genetic Clinics |
| Editorial board |
Get Our Newsletter |
| Subscribe |
Send Your Feedback |
| Feedback Form |
About Us |
| IAMG |
GeNeViSTA
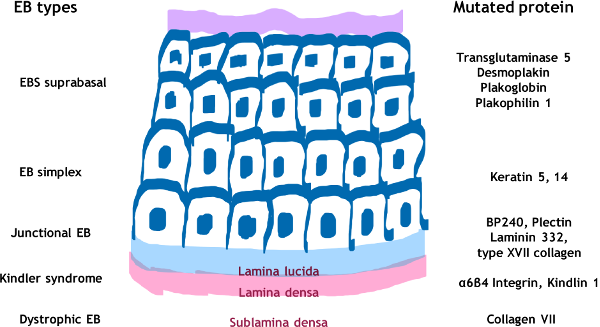
| Features | EB simplex | Junctional EB | Dystrophic EB | Kindler syndrome |
| Skin fragility | + (with little or no trauma) | + (with little or no trauma) | + (with little or no trauma) | + (with little or no trauma) |
| Blisters |
|
|
|
|
| Age | Birth-12-18 months* | Birth | Birth | Birth |
| Distribution | Hands and feet (Localized form)/generalized | Periorificial areas, fingers and toes, trunk and the upper airway mucosa | Hands, feet, knees, and elbows (mild forms)-whole body including mucosa (in severe forms)** | Acral |
| Characteristic Pattern | Annular or curvilinear groups or clusters | Excessive periorificial granulation tissue | Localized or generalized with scarring and milia formation |
|
| Mucosal | + (generalized severe forms) | + (oral and airway mucous membrane**) | + (severe forms) | + |
| Post inflammatory changes | + hyper/hypo pigmentation (generalized forms) | + hyper/hypo pigmentation | + scarring | + skin atrophy |
| Skin scarring | No | -/+ (generalized severe form) | + (scarring, pseudosyndactyly, “mitten” hands and feet & contractures) | + |
| Nail dystrophy | -/+ (generalized severe forms) | -/+ | + and nail loss especially toenails | -/+ |
| Milia | -/++ (generalized severe forms) | - | + | - |
| Others | Varying degree of palmar and plantar hyperkeratosis (generalized severe forms) | Prone to sepsis, amelogenesis imperfecta, congenital malformations of the urinary tract and bladder, aplasia cutis, non-scarring or scarring alopecia | Oral and/or esophageal scarring and strictures. Corneal erosions resulting in corneal opacity leading to loss of vision. High risk of squamous cell carcinoma | Esophageal stricture, photosensitivity, telangiectasia, colitis, urethral stenosis/strictures, and severe phimosis |
| Inheritance | AD/AR | AR | AD/AR | AR |
EB – Epidermolysis bullosa; AD – Autosomal dominant; AR – Autosomal recessive
* Varies depending upon the severity, gene and the variant
** Mucosal involvement of the mouth, upper respiratory tract, esophagus, bladder, urethra, and corneas
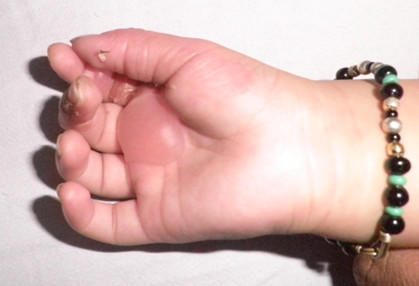
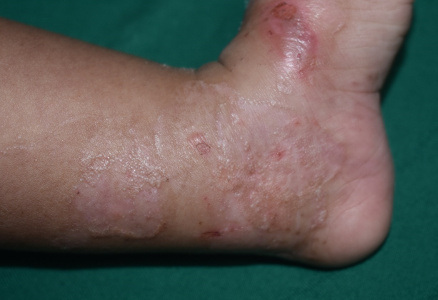
In EB simplex localized, blisters are late in onset and are mostly localized to trauma prone areas (Figure 2). Generalized severe form of EB starts at birth resulting in extensive and grouped blistering resolving with hypo or hyperpigmentation (Figure 3). Palmoplantar keratoderma is common and begins in childhood. Nail dystrophy and milia can be seen. The blistering is less severe in the generalized intermediate form of EBS.
Various genes implicated in various subtypes of EBS are shown in Tables 2 and 3. Most cases of EBS are caused by mutations in KRT5 and KRT14 (keratin 5 and 14) genes (Pfendner and Bruckner, 1998). EBS patients with mutation in the TGM5 (transglutaminase 5) gene have mild blistering of skin over the acral areas consistent with the diagnosis of acral peeling syndrome. Biallelic mutations in the DSP (desmoplakin) gene result in lethal acantholytic EB simplex characterized by skin fragility, universal alopecia, malformed ears, anonychia and cardiomyopathy. Mutation in the EXPH5 (exophilin-5) and KRT14 genes causes mild form of EBS with an autosomal recessive inheritance. Biallelic mutations in PLEC (plectin) gene are known to have EBS with associated muscular dystrophy and relatively higher morbidity and systemic complications. Site-specific monoallelic missense mutation in PLEC gene results in EBS Ogna type (EBS-O). Patients with EBS with migratory circinate erythema and mottled pigmentation have been found to have mutation in KRT5. There are also reports of EBS with mottled pigmentation caused by mutation in KRT14. Recently, some patients of EBS have been shown to have mutation in KLHL24 gene. Digenic inheritance with mutations in KRT5 and KRT14 has also been described.
| Subtype | Gene |
| Acral peeling skin syndrome (APSS) | TGM5 (Transglutaminase 5) |
| Epidermolysis bullosa simplex superficialis (EBSS) | - |
| Acantholytic Epidermolysis bullosa simplex (EBS-acanth) | DSP (Desmoplakin) |
| Skin fragility syndromes:
|
DSP
(Desmoplakin) |
| Subtype | Gene |
| Major subtypes:
Others:
|
KRT5
and
KRT14
(Keratins
5
and
14)
PLEC
(Plectin) |
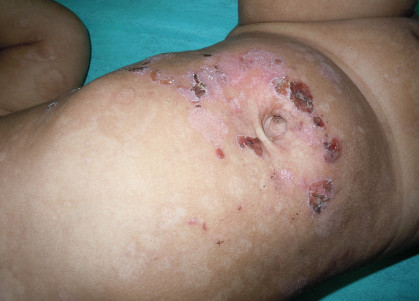
Generalized (Herlitz) severe junctional EB is the most devastating type of EB resulting in severe life-threatening complications in early childhood. Extensive truncal erosions at the time of birth and periorificial granulation tissue is characteristic of this form of EB (Figures 4a and b). A series of junctional EB showed that 73% of 71 children born in a five-year period died at an average age of five months (Kelly-Mancus et al., 2014). The common causes of death are sepsis, electrolyte imbalance, renal and cardiac complications. Accumulation of granulation tissue in subglottic area in these children results in weak hoarse cry. Eventually airway obstruction may result in stridor and respiratory distress and tracheostomy is required. Amelogenesis imperfecta with pitting of tooth enamel and scarring or non-scarring alopecia are common. Generalized intermediate type is a less severe clinical presentation of JEB where blistering may be localized to trauma-prone areas and systemic complications are rare.
Table 4 shows various genes involved in JEB. Junction EB is most commonly caused by mutations in genes coding for alpha-3, beta-3, and gamma-2 subunits of laminin protein of lamina lucida of the basement membrane zone. The lethal Herlitz type of junctional EB is known to be caused by premature termination or missense biallelic mutations in LAMB3 (laminin B3) gene while junction EB intermediate is caused by biallelic mutations in the COL17A1 gene (Yenamandra et al., 2017). Laryngo-onycho-cutaneous syndrome (LOCS) characterized by granulating wounds, dental hypoplasia and ocular granulation tissue is known to be caused by biallelic mutations in the LAMA3 gene. Junctional EB with pyloric atresia (EB-PA) is caused by biallelic mutations in ITGA6 or ITGB4 genes that code for α6β4 integrin in the basement membrane zone (Kayki et al., 2017).
| Junctional EB type | Junctional EB subtype | Targeted protein(s) |
| Generalized | ∙ Severe | ∙ Laminin 332 |
|
| ∙ Intermediate | ∙ Laminin-332, type XVII collagen |
|
| ∙ Late onset | ∙ Type XVII collagen (COL17A1) |
|
| ∙ With pyloric atresia | ∙ α6 (ITGA6) and β4 integrin (ITGB4) |
|
| ∙ With respiratory (interstitial lung disease) and renal involvement (nephrotic syndrome) | ∙ α3 integrin subunit (ITGA3) |
| Localized | ∙ Localized | ∙ Laminin-332, type XVII collagen, α6 and β4 integrin subunits |
|
| ∙ Inversa | ∙ Laminin-332 |
|
| ∙ Laryngo-onycho-cutaneous (LOC) syndrome | ∙ Laminin α3a (LAMA3A) |
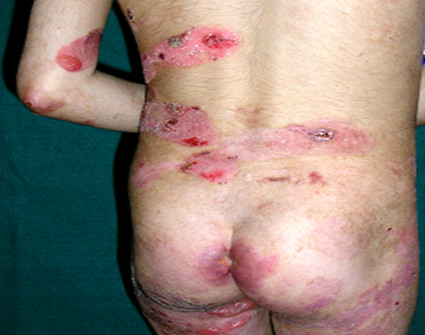
Generalized recessive dystrophic EB (RDEB) results in extensive blistering and erosions at birth resolving with milia and scaring while dominant dystrophic EB (DDEB) is a milder form of the disease where the disease is mostly limited to trauma-prone areas associated with nail dystrophy (Figures 5a, b and c). Presence of syndactyly, mitten like deformity, systemic complications, dental lesions, remission-less course, and oral lesions are strongly indicative of RDEB (Yenamandra et al., 2017). Esophageal erosions and stricture formation can lead to severe dysphagia and resultant malnutrition in these patients. Corneal erosions can lead to corneal opacity and loss of vision. The non-healing wounds of dystrophic EB may be complicated by aggressive squamous cell carcinoma.
Dystrophic EB results from mutation in COL7A1 (collagen type 7) gene and more than 300 pathogenic variations have been detected in this gene causing various subtypes of dystrophic EB (Dang et al., 2008).
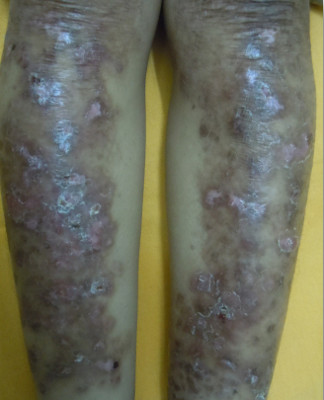
EB pruriginosa is a distinct variant of dystrophic EB characterized by severe pruritic lichenified plaques over bilateral lower legs associated with nail dystrophy and is caused by mutation in the COL7A1 gene (Figure 6). Pretibial form of dystrophic EB is characterized by blistering, milia and scarring limited to the pretibial area associated with dystrophic nails. It is caused by mutation in the NC2 domain of COL7A1. Bullous dermolysis of newborn is a self-limiting disease caused by mutation in COL7A1 and manifests as generalized bullae and erosions in the newborn period resolving by itself by one year of age. Inversa recessive dystrophic EB (RDEB-I) is a rare form, characterized by generalized involvement in the neonatal period which improves with age and a predominant involvement of flexures is seen in adults. Severe mucosal involvement is seen in these cases. A late onset of inversa RDEB is also reported. Dominant DEB presenting only as familial nail dystrophy has been reported caused by mutation in COL7A1.
| Dystrophic EB type | Dystrophic EB subtype | Targeted protein(s) |
| Dominant dystrophic epidermolysis bullosa (DDEB) | DDEB, generalized DDEB, acral DDEB, pretibial DDEB, pruriginosa DDEB, nails only DDEB, bullous dermolysis of newborn | Collagen 7 |
| Recessive dystrophic epidermolysis bullosa (RDEB) | RDEB, generalized severe type RDEB, generalized other RDEB, inversa RDEB, pretibial RDEB, pruriginosa RDEB, centripetalis RDEB, bullous dermolysis of newborn | Collagen 7 |
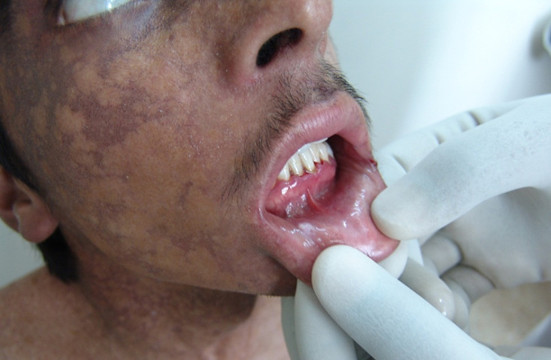
Kindler syndrome is characterized by acral blisters early in life followed by poikiloderma, periodontitis and involvement of oral, oesophageal, urogenital and ocular mucosa (Figure 7) (Kantheti et al., 2017).
| Type | Targeted protein(s) | Gene |
| Kindler syndrome | Kindlin 1 | FERMT1 |
In most patients diagnosis of EB is made clinically based on the presence of characteristic clinical findings (Yenamandra et al., 2017).
Immunohistochemistry studies: Diagnostic of specific EB type requires identification of the level of split via antigen mapping on a newly induced blister. Antigen mapping refers to the use of monoclonal antibodies in immunofluorescence or immunohistochemistry directed against components of the basement membrane zone and detecting their altered staining corresponding to the presence of a mutation in their gene (Yenamandra et al., 2017).
Molecular genetic testing: The gold standard for the diagnosis is confirmation by DNA analysis using next generation sequencing, if available and affordable, as this will permit the identification of the specific EB subtype, help in detection of newer types, and in prenatal genetic diagnosis, genetic counselling and future gene therapy. The different molecular testing approaches are single gene testing, use of a customized multi-gene panel or a whole exome analysis. Epidermolysis bullosa sequencing based diagnostic assay called EBSEQ was developed recently for simultaneous detection of 21 genes with a known role in EB (Lucky et al., 2018).
The definitive treatment for EB is not available yet and mainstay of management is supportive care.
Supportive treatment: Avoidance of trauma, proper wound care to prevent secondary infection and scarring and to promote early healing, nutritional support, genetic counseling and management of complications remains the primary treatment of disease. Recently, a wound care guideline was compiled by international experts (Pope et al., 2012). These guidelines recommended adequate wound assessment and use of appropriate wound dressing according to the wound type. Bath soaks are recommended prior to removal of wound dressings to minimize the trauma. Addition of salt to bath water has shown to relieve the pain and prevent infection. The frequent use of antibiotics is discouraged to prevent development of antibiotic resistance in these patients. Adequate management of pain and pruritus is recommended to improve the quality of life. Assessment of nutritional deficiency and its correction is very important for the proper growth and development of these children. Physical therapies to improve the mobility and use of aids to improve their functionality are also essential for their well-being.
Newer therapies: Newer experimental therapies include gene therapy, fibroblast and protein therapy and mesenchymal and bone marrow transplantation (Gostyńska et al., 2018; Hirsch et al., 2017). Extensive research and long-term studies are required to evaluate the efficacy and safety of these experimental therapies.
DEBRA (Dystrophic Epidermolysis Bullosa Research Association) International (http://www.debra-international.org/homepage.html) is an umbrella organization for a worldwide network of national groups that work for children affected with epidermolysis bullosa. The organization provides guidelines and support for better research and care of these children.
1. Dang N, et al. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol 2008; 17: 553-568.
2. Fine JD. Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates from the National Epidermolysis Bullosa Registry. JAMA Dermatol 2016; 152: 1231-1238.
3. Fine JD, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014; 70: 1103-1126.
4. Gostyńska KB, et al. Allogeneic Haematopoietic Cell Transplantation for Epidermolysis Bullosa: the Dutch Experience. Acta Derm Venereol 2019; 99: 347-348.
5. Hirsch T, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017; 551: 327-332.
6. Kantheti P, et al. Two novel mutations in KIND1 in Indian patients with Kindler syndrome. Clin Exp Dermatol 2017; 42: 95-97.
7. Kayki G, et al. Epidermolysis Bullosa with Pyloric Atresia and Aplasia Cutis in a Newborn Due to Homozygous Mutation in ITGB4. Fetal Pediatr Pathol 2017; 36: 332-339.
8. Kelly-Mancuso G, et al. Junctional epidermolysis bullosa incidence and survival: 5-year experience of the Dystrophic Epidermolysis Bullosa Research Association of America (DebRA) nurse educator, 2007 to 2011. Pediatr Dermatol 2014; 31:159-162.
9. Liu L, et al. Autosomal recessive epidermolysis bullosa simplex due to loss of BPAG1-e expression. J Invest Dermatol 2012;132 (3 Pt 1):742-744.
10. Lucky AW, et al. A comprehensive next-generation sequencing assay for the diagnosis of epidermolysis bullosa. Pediatr Dermatol 2018; 35:188-197.
11. Pfendner EG, Bruckner AL. Epidermolysis bullosa simplex. In: GeneReviews® [Internet]. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. Seattle (WA): University of Washington, Seattle; 1993-2019.
12. Pope E, et al. A consensus approach to wound care in epidermolysis bullosa. J Am Acad Dermatol 2012; 67: 904-917.
13. Yenamandra VK, et al. Development of a clinical diagnostic matrix for characterizing inherited epidermolysis bullosa. Br J Dermatol 2017;176: 1624-1632.
14. Yenamandra VK, et al. Application of whole exome sequencing in elucidating the phenotype and genotype spectrum of junctional epidermolysis bullosa: A preliminary experience of a tertiary care centre in India. J Dermatol Sci 2017; 86: 30-36.
15. Yenamandra VK, et al. Diagnosis of Inherited Epidermolysis Bullosa in Resource-Limited Settings: Immunohistochemistry Revisited. Dermatology 2017; 233: 326-332.
| Abstract | Download PDF |
 a)
a) 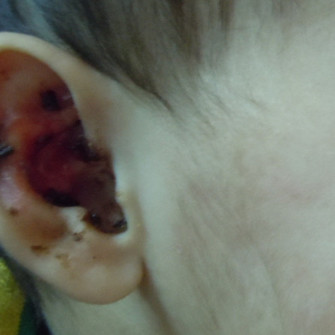 b)
b)
 a)
a) 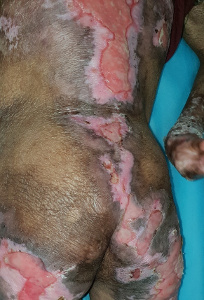 b)
b) 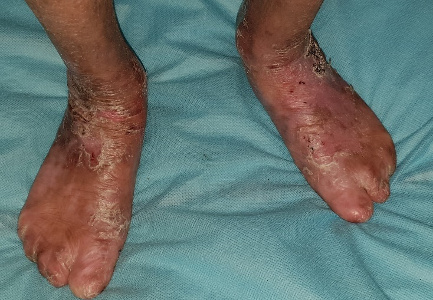 c)
c)