Mosaicism in Clinical Genetics: Counselling Challenges and Diagnostic Dilemmas
Haseena Sait Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India Correspondence to: Dr Haseena SaitEmail:hasi.flower@gmail.com
Abstract
Mosaicism involves the presence of more than one genetically distinct cell line within a single organism,
contributing to a range of pathologies from chromosomal disorders to various cancers. In clinical practice,
mosaicism presents significant challenges, from suspicion to detection. Equally complex is the process of genetic
counselling for mosaic disorders. This review focuses specifically on non-oncological conditions related to
mosaicism. It outlines the clinical classification, mechanisms, and common presentations of mosaic conditions.
Additionally, the article provides an overview of mosaicism in different clinical settings, including various
diagnostic techniques and the uncertainties and challenges in genetic counselling related to mosaicism.
Keywords: Germline mosaicism, somatic mosaicism
Introduction
Mosaicism is defined as the occurrence of two or more cell lines with a different genetic composition derived from a single
fertilised egg within a single individual. Mosaicism can have a wide range of effects, from early pregnancy loss to organ
specific pathology, to modification of clinical syndromes.
Classification
The developmental timing and cell lineage affected, along with the phenotypic consequences of the mutation, ultimately
determine the tissue and cell type distribution of mosaicism and also the patterns of disease recurrence within families.
The broad clinical classification includes:
Somatic- occurring only in the cells of the body, but not including the germline.
Germline- occurring only in the germ cells or their precursors but not found elsewhere in the body.
Mixed gonadal and somatic(Gonosomal)- occurring in both the cells of the body and the germline.
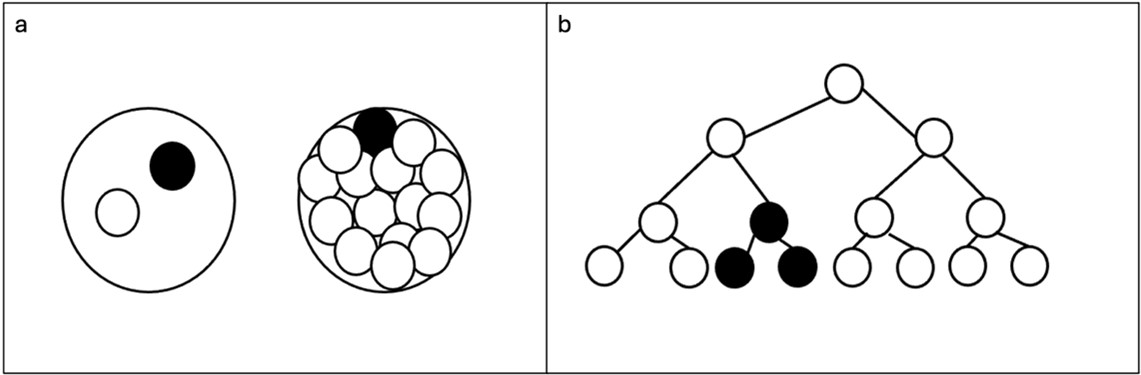
Figure 1: a) De novo postzygotic genetic alteration (dark circle) in two cell or multicellular stage of zygote b) De
novo postzygotic genetic alteration (dark cell) in an actively dividing cell
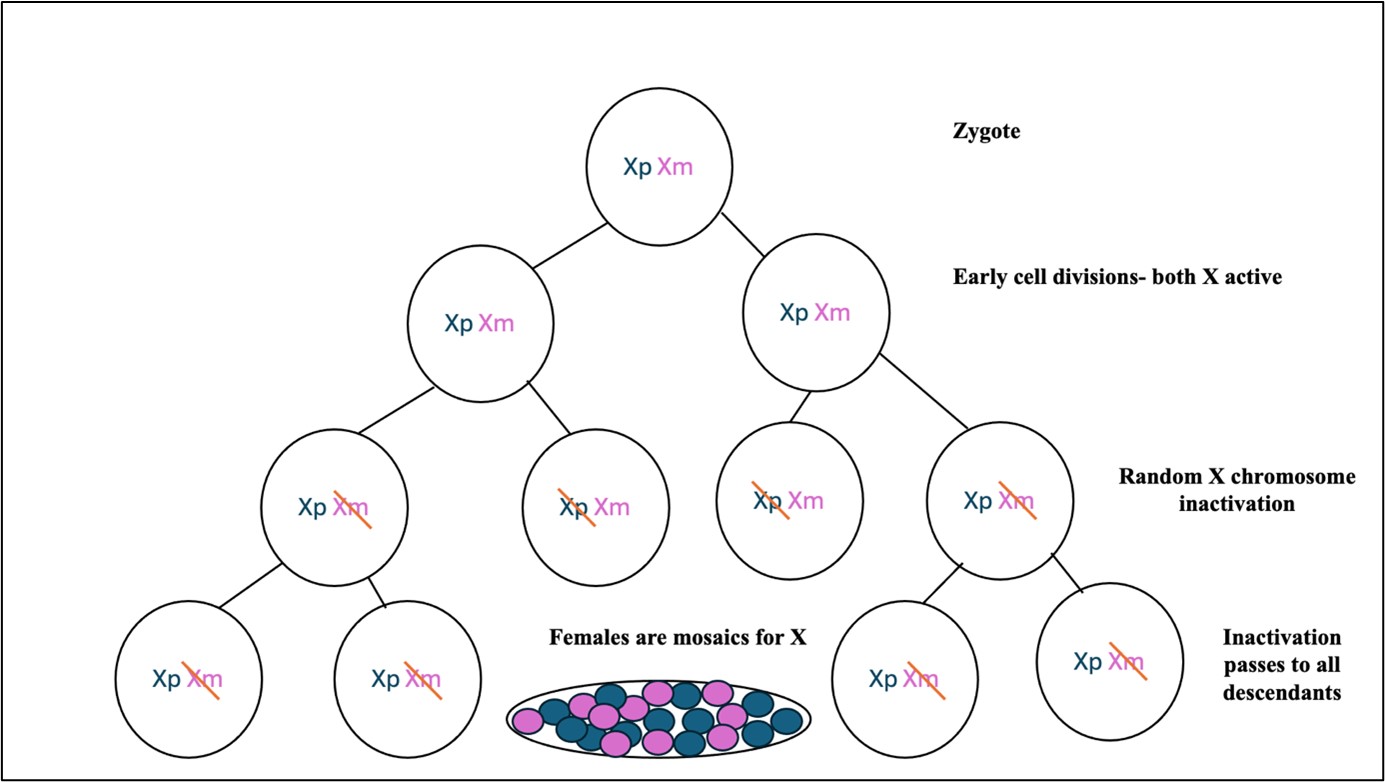
Figure 2: Random inactivation of one of the X chromosome in females through epigenetic silencing Xp: paternallyderived X chromosome, Xm: maternally derived X chromosome
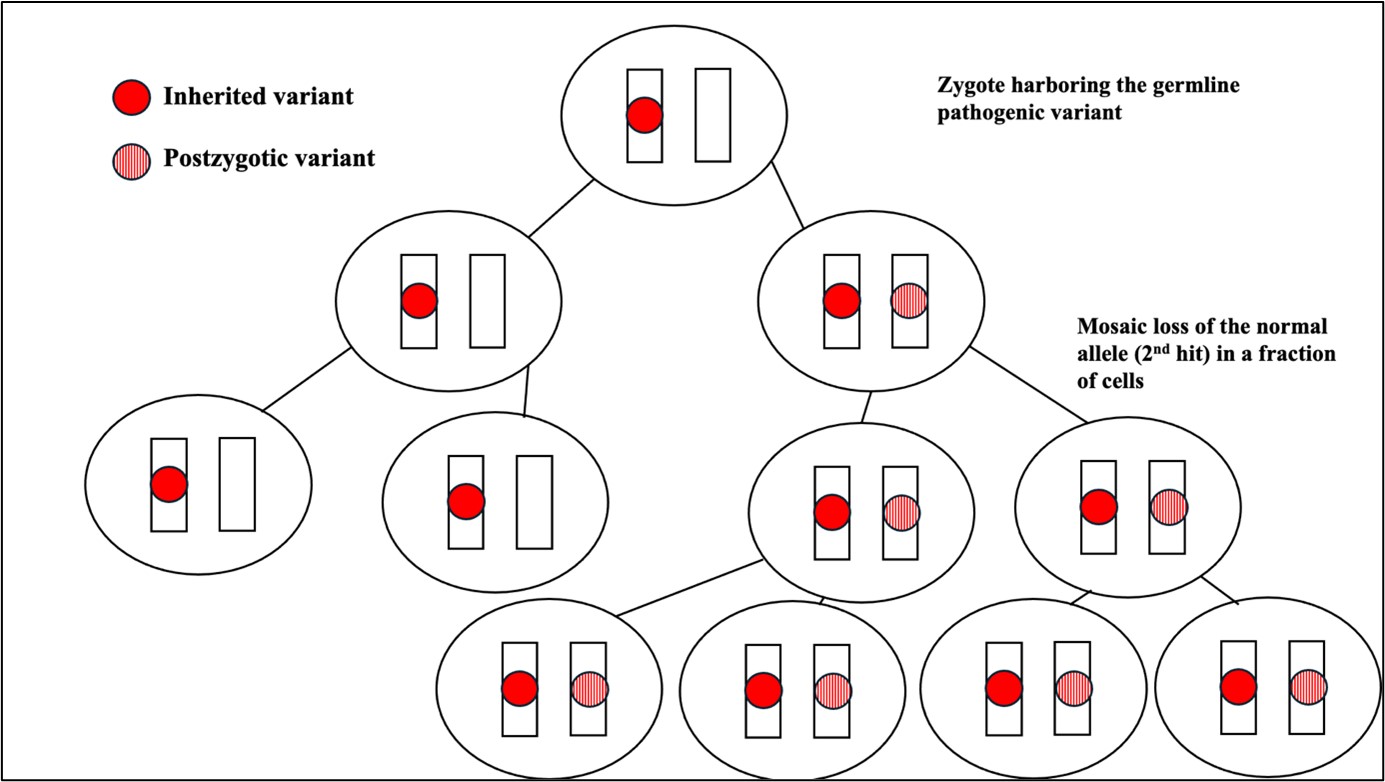
Figure 3: Mosaic nullizygosity- 2nd hit (genetic alteration) in a fraction of cells which already carry a germline
variant
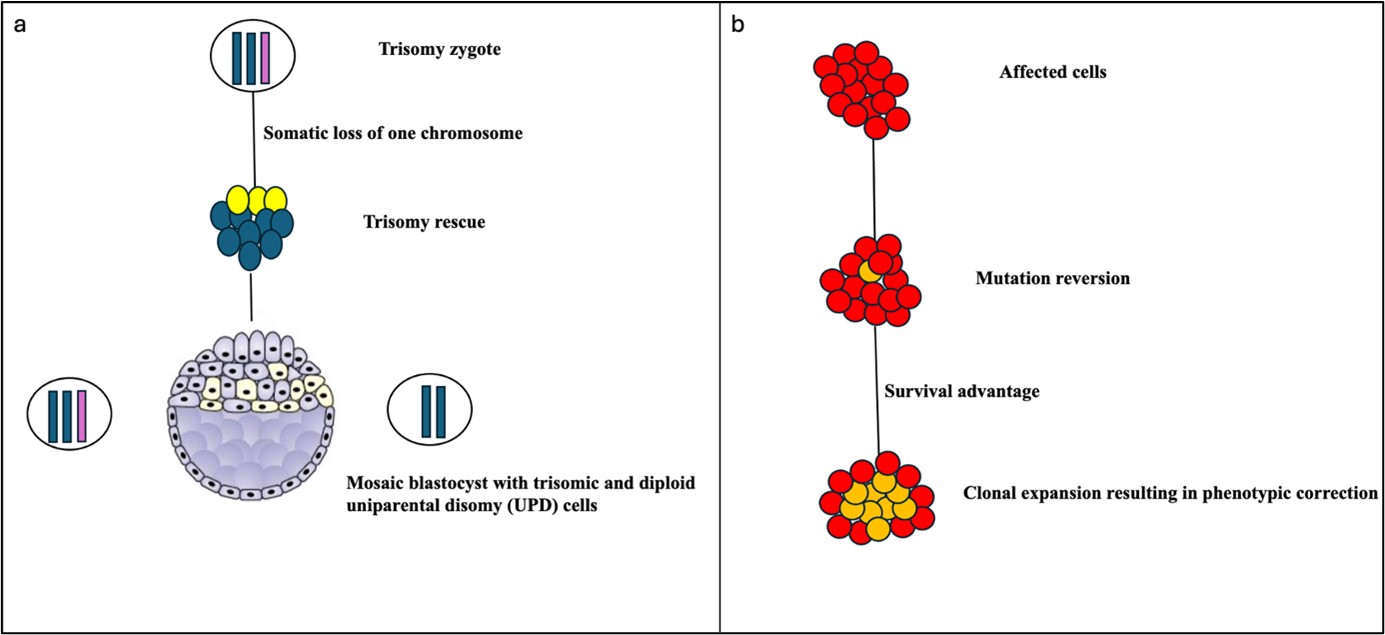
Figure 4: a) Mosaic blastocyst due to trisomy rescue, b) Revertant Mosaicism
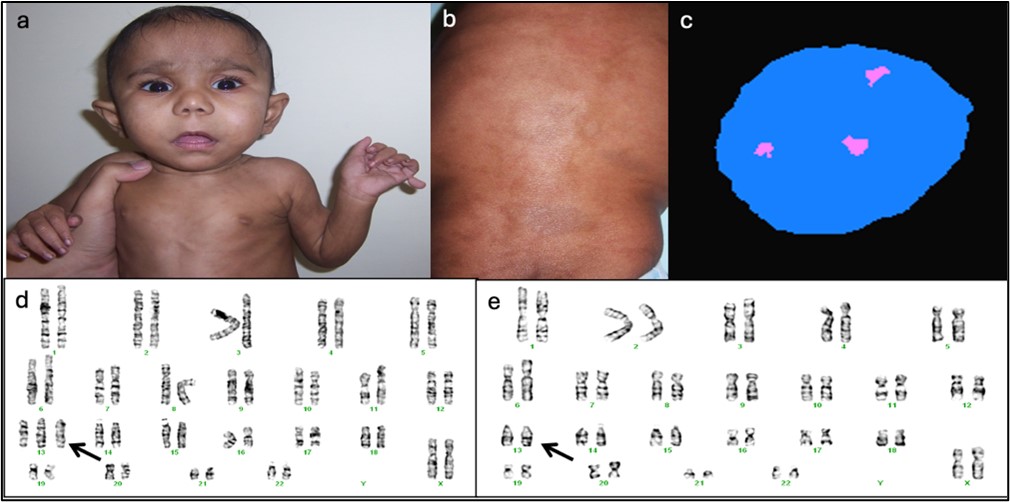
Figure 5: a) Child with global developmental delay and postaxial polydactyly, b) Patchy pigmentary abnormality
of skin over back, c) Interphase FISH: 46,XX,+13 [80] / 46,XX [20], d and e: Karyotype: 47,XX,+13 [13] /
46,XX[7]. Picture courtesy: Dr Shubha R Phadke
Mechanisms causing mosaicism (Wallace et al., 2022)
De novo postzygotic genetic alteration(chromosomal or single nucleotide variation): This can occur
in the embryo any time after the first cell division (Figure 1a) or in an actively dividing cell throughout
the life of an individual resulting in somatic, germline, and/or placental mosaicism (Figure 1b).
Epigenetic silencing: This causes inactivation of select genes on the X chromosome in cells with more
than one X chromosome. E.g., females are natural mosaics for X chromosome (Figure 2).
Mosaic nullizygosity: A mosaic (or postzygotic) genetic alteration occurring in an actively dividing cell
in trans with a germline pathogenic variant resulting in mosaic loss of the normal allele in a fraction of
cells. This causes type 2 segmental mosaicism as seen in conditions like neurofibromatosis type 1, tuberous
sclerosis (Figure 3).
Rescue mechanisms:
Spontaneous postzygotic loss of a trisomic chromosome resulting in mosaic uniparental disomy
(Figure 4a)
Spontaneous reversion of a germline genetic alteration in a dividing cell that eliminates the germline
variant (i.e., revertant mosaicism) (Figure 4b).
Common clinical presentations of mosaic disorders:
Mosaicism tends to present with the following clinical manifestations:
Patchy distribution of pigmentary lesions (linear or whorled hyper- and/or hypopigmentation) is an
important clinical clue. It may not be always obvious on external examination. For a few disorders like
intellectual disability, the presentation can be mild and often overlooked.
Growth abnormalities especially causing asymmetric, focal or segmental involvement.
Chromosomal mosaicism (Pre and postnatal scenarios)
Mosaicism for many types of chromosome abnormalities is well known in both abortuses and liveborn
individuals. A constitutional gain or loss of only a few single chromosomes is compatible with viability:
trisomy of chromosomes 13, 18, 21 and X. All of these trisomies or monosomies have also been detected as
mosaic abnormalities, usually with a milder phenotype than the constitutional aneuploidy (Figure 5).
Postnatally, the presence of patchy pigmentary abnormalities is a clue to possible chromosomal mosaicism.
The presence of a normal cell line in patients with common trisomies, such as trisomy 21 or trisomy 18,
may suggest better cognitive function, but there is no correlation between the level of mosaicism and the
outcome.
An additional class of aneuploidies can be found only when they are mosaic, presumably due to selection against the
aneuploid cells in a specific tissue or at a specific stage of development. These include trisomies of chromosomes 8, 9, 14,
17, and 22.
Mosaicism for various types of structural abnormalities has also been identified, including balanced and unbalanced
translocations, deletions, duplications, inversions, ring chromosomes and isochromosomes (Biesecker et
al., 2013). Isochromosomes are often supernumerary, presumably because the loss of even one copy of a
chromosome arm can be lethal with a common exception of isochromosome Xq causing Turner syndrome which is
well tolerated. Some of the commonly recognisable syndromes associated with isochromosomes include
isochromosome 12p (Pallister– Killian syndrome), isochromosome 22q (cat eye syndrome), isochromosome 15q11
and isochromosome 18p.The isochromosome 12p seen in Pallister–Killian syndrome is always present in a
mosaic form, as the abnormality would be lethal when constitutional. The mosaicism in these patients is
almost always limited to cultured fibroblasts and is usually not seen in lymphocyte cultures. However, it
can be detected sometimes with cytogenetic microarray techniques using DNA extracted from peripheral
blood.
Prenatally, the effects of mosaic chromosomal aneuploidies on fetuses depend on the type of chromosome involved,
mosaicism levels, and the tissues involved, the intrauterine phenotype and clinical outcomes. UPD and
chromosomal aneuploidy often co-occur, so it is necessary to analyze them simultaneously when chromosomes with
imprinted regions (chromosomes 6, 7, 11, 14, 15, 20) are involved or when there are known carriers of a
recessive allele. The information provided during genetic counseling should include the affected chromosomes,
mosaicism levels, mosaic tissues involved, methylation status, recessive gene variations on the UPD chromosome,
intrauterine phenotypes, clinical manifestations of similar patients after birth, and current clinical treatments
available.
Mosaic manifestations of Mendelian disorders
The mosaic manifestations of Mendelian disorders can be divided into three groups:
Mosaicism for lethal mutations causing clinical pictures that exist only in mosaic form
Mosaicism for mutations known in autosomal-dominant/X linked recessive disorders
Rare mosaicism that causes aggravation of the phenotype in a segmental area due to a second mutation
event on the other allele (usually loss of heterozygosity) in autosomal-dominant inherited disorders
Mosaicism for lethal mutations causes clinical pictures that exist only in mosaic form
Some mosaic disorders are caused by mutations that are seen only in mosaic form. These disorders
are lethal in its constitutional state and hence cannot be passed on by the affected individuals to their
offsprings. The classic example for this group of disorders is McCune–Albright syndrome (MAS) that occurs
due to mosaic gain-of-function mutations in the GNAS1 gene. Other classic examples include overgrowth
disorders that are caused by activating mutations in PIK3CA, AKT3 and MTOR pathways (Moog et al.,
2020).
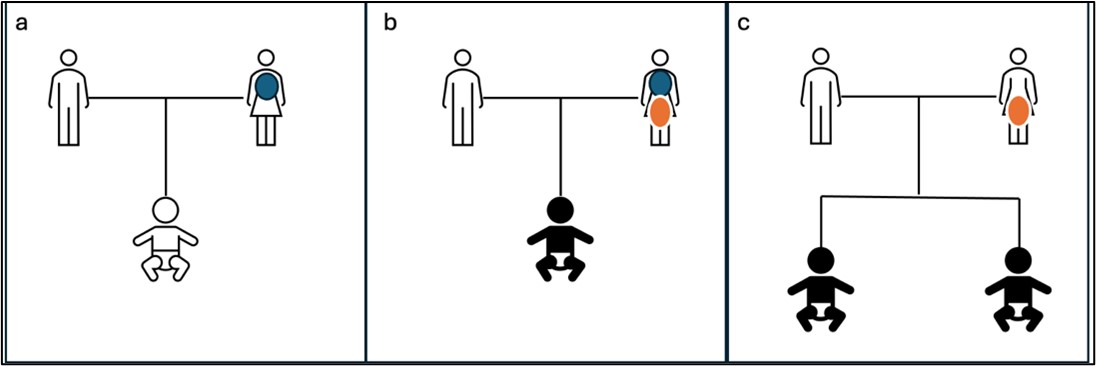
Figure 6: a: Mother carries mosaic variant in somatic cells and is mildly affected. Child is phenotypically normal.
b: Mother carries mosaic mutation in both somatic and gonadal cells. Mother is mildly affected but child is
affected with a generalized and severe form of the disease. c: Mosaic variant is restricted to gonadal cells in mother.
Mother is phenotypically normal but multiple children are affected. Blue circle: Somatic mosaicism, Orange circle:Gonadal mosaicism
Reproductive counselling These disorders are not inherited and arise sporadically early in embryonic development. The risk for an affected sibling is
expected to be the same as in the general population. There is no possibility of vertical transmission due to embryonic
lethality.
Mosaicism for mutations known in autosomal-dominant/X-linked recessive disorders
Some of the common monogenic disorders for which somatic/ gonadal mosaicism is well documented include Duchenne
muscular dystrophy, osteogenesis imperfecta, neurofibromatosis, hemophilia, Marfan syndrome etc. Somatic
mosaicism is reported in 25-33% and 6.5% of persons with neurofibromatosis type 2 and type 1 respectively
(Ruggieri and Huson., 2001). Somatic/gonadal mosaicism is present in 16% of asymptomatic parents of
autosomal dominant COL1A1 and COL1A2 osteogenesis imperfecta patients (Pyott et al., 2011). Somatic
mosaicism is reported in as high as 43% of persons with ADCY5 related dyskinesias (Raskind et al., 2017).
Some diseases, such as Cornelia de Lange syndrome (CDLS), show markedly high rates of mosaicism, even
across mutational mechanisms and genomic loci (somatic mosaicism is reported in 10-15% of persons with
NIPBL-related CDLS; Huisman et al., 2013). Mosaicism for increased APP gene copy number has been identified in
the brains of individuals with Alzheimer’s dementia, suggesting that mosaicism for mutations that cause
Mendelian disease when present constitutionally may also contribute to sporadic disease (Bushman et al.,
2015).
The severity and clinical symptoms of postzygotic mosaicism depend on the timing of the mutation event,
the type of cell in which the mutation occurs, the expansion of cells with mutations, the mutated gene,
and the specific mutation. Depending on the timing of the mutation event, these mosaics can present in a
disseminated manner, causing atypical or attenuated forms of a clinical picture, or localized as segmental
mosaicism type I, which generally has milder effects, such as in segmental neurofibromatosis type 1 (NF1) or
mosaic forms of tuberous sclerosis. Parents with segmental neurofibromatosis can produce offspring with a
constitutional/generalized form of NF1 when gonosomal mosaicism underlies their disease (Consoli et al., 2005).
Sometimes, mosaicism can result in phenotypes that differ significantly from the expected manifestation of the disorder,
potentially presenting as a different disorder altogether. For example, mosaic RASopathies have a different
phenotype compared to constitutional RASopathies, being based on postzygotic gain-of-function mutations in
RAS/RAF genes, which would presumably be lethal if they were constitutional. (Hafner and Grosser.,
2013)
Reproductive Counselling Parents with somatic mosaicism can produce offspring with a constitutional form of the disorder if their gonadal
tissue is also affected. On the other hand, pure germline mosaicism is typically identified when multiple
siblings are born with a genetic condition, despite their parents being unaffected (Figure 6). This occurs
because the mutation arises during the formation of the parent's germline cells, meaning it is confined
to the germline and not present in the parent's somatic cells, leaving them asymptomatic. Since genetic
testing in parents usually examines DNA from blood samples, it would generally reveal the wild-type variant
in such cases. Although there is empirical data on recurrence risks for some sporadic forms of autosomal
dominant or X-linked disorders, this information is lacking for many rare de novo conditions. Therefore,
prenatal testing is currently justified for all families with sporadic cases of autosomal dominant or X-linked
conditions.
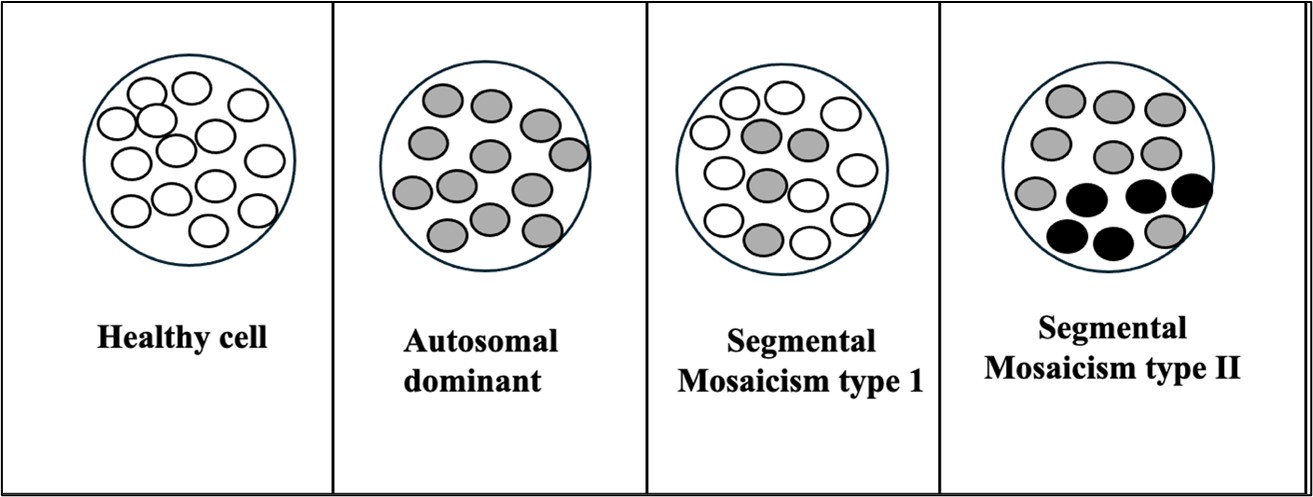
Figure 7: Segmental Mosaicism. Type 1: mosaic mutation in subset of cells, Type 2: second mutation in the
subpopulation of precursor cells in an individual who already carries a germline mutation White circle: Wild allelein homozygous state, Grey circle: Mutated allele in a heterozygous state, Dark circle: Two mutated alleles
Rare mosaicism that causes aggravation of the phenotype in a segmental area
Type II segmental mosaicism is a type of segmental mosaicism in which a second mutation occurs in the subpopulation
of precursor cells in an individual who already carries a germline mutation. This leads to segments with more severe
phenotypes. Examples of type II segmental mosaicism are manifestations of Hailey-Hailey and Dariers diseases with
stripes of more severe cutaneous disease (Figure 7).
Epigenetic Mosaicism
Epigenetic mosaicism is defined as the presence of different epigenotypes in different cell populations within an organism
developed from a single zygote. Epigenetic mosaicism is described in a number of genomic imprinting disorders, including
hydatidiform mole, Angelman syndrome (AS), Prader–Willi syndrome (PWS), Silver–Russell syndrome (SRS),
Beckwith–Wiedemann syndrome (BWS), Temple syndrome, pseudohypoparathyroidism 1B, and transient neonatal
diabetes mellitus (TNDM). Epigenetic mosaicism in genomic imprinting disorders seems to have different contributions to
the formation of the clinical picture of the disorder. It was demonstrated that mosaicism in the case of BWS, PWS, and
AS was associated with a milder phenotype and the appearance of clinical features were not characteristic of the given
pathology (Sazhenova and Lebedev., 2019). The leading diagnostic features of AS include gait ataxia and absence of
speech; whereas patients with SNURF-SNRPN imprinting centre mosaicism are able to speak single words, they have no
stereotypic movements, and in some cases, there are clinical features overlapping with PWS (obesity and hyperphagia)
(Le-Fevre et al., 2017). Mosaicism can therefore significantly obscure the typical clinical presentation of syndromes,
making both clinical diagnosis and molecular genetic analysis challenging. For disorders like TNDM and
SRS, the classical phenotype can result from even a small clone of epigenetically altered cells. In addition,
epigenetic mosaicism was observed only in certain tissues in patients with TNDM (Sazhenova and Lebedev.,
2019).
Fragile X syndrome is a triplet repeat disorder often characterized by the presence of mosaicism in FMR1 variants.
This mosaicism can manifest as either repeat size mosaicism or methylation mosaicism. Studies indicate that individuals
with methylation mosaicism tend to perform at a higher intellectual level compared to those with fully methylated full
mutations. This is because the production of FMR1 protein (FMRP) occurs from the transcription of the unmethylated
alleles, which supports better cognitive functioning.
Revertant and Rescue Mosaicism
Revertant mosaicism is a phenomenon in which a mutation gets spontaneously corrected in a subset of cells in an affected
organ perhaps driven by selective pressure. Revertant mosaicism can occur in the germline or in somatic cells. Invivo reversion of somatic cells tends to predominantly involve tissues with high cell proliferation rates
including the skin, liver and the hematopoietic system (Lai-Cheong et al., 2011). The classic examples include
epidermolysis bullosa, ichthyosis, adenosine deaminase deficiency, Wiskott-Aldrich syndrome, Bloom syndrome,
Fanconi anemia, tyrosinemia I, etc. Germline revertant mosaicism has previously been described in myotonic
dystrophy wherein the size of the CTG repeats in two unrelated healthy individuals, born to clinically affected
parents, was normal despite having inherited the myotonic dystrophy DNA-marker haplotype (Brunner et al.,
1993)
Revertant mosaicism often leads to phenotypic improvement and is therefore called “natural gene therapy”. It can also
modify the phenotypic expression leading to atypical presentations of disease (Wada et al., 2008). The timing of reversion
during development can also influence the extent of revertant mosaicism and the severity of the condition. Very rarely,
unfavourable outcome due to overcorrection of mutants by somatic mosaicism has also been reported (Inaba and
Nagamachi., 2021)
Confined placental mosaicism
Confined placental mosaicism (CPM) is defined as a chromosomally abnormal cell line restricted to the placenta, while the
chromosomes of the fetus itself are normal. It arises either due to non-disjunction in a diploid conception as a
post-zygotic error or by a trisomic rescue mechanism, wherein a viable trisomic conceptus loses one chromosome
through anaphase lagging and produces a diploid cell line. CPM can be categorized into three subtypes (type
1, 2 and 3) depending on where the chromosomal abnormality is found in the placenta (Toutain et al.,
2018).
Type 1 : In type I CPM, the chromosomal abnormality is only found in the cytotrophoblast and can be found after
examination of the short-term culture villi. Nowadays, this methodology is not in use as rapid results are available with
other techniques like fluorescence in situ hybridization (FISH) and quantitative fluorescent polymerase chain reaction
(QFPCR) for detection of aneuploidies.
Type 2 : In type 2 CPM, the chromosomal abnormality is only found after long-term culture of villi and is restricted
to the mesenchymal core of the chorionic villi.
Type 3 : Type 3 CPM is characterized by the presence of the abnormality in both the mesenchymal core and
cytotrophoblast and can be found after both long term and short-term cultures.
CPM is believed to occur in roughly 1–2% of all placental tissue analysis. The presence of chromosomally abnormal
cells restricted to the placental areas that are not sampled by chorionic villus sampling (CVS) can go unnoticed.
Non-invasive prenatal screening on the other hand appears to be more sensitive to detect CPM as compared to CVS, as
the entire placental trophoblast sheds cfDNA into the maternal circulation (Brison et al., 2018; Van Opstal et al.,
2018)
CPM significantly contributes to fetal growth restriction (FGR) (71.7%) and is associated with a high
rate of premature births (31%). It is crucial to conduct thorough examinations of CPM pregnancies for
structural fetal anomalies, given the notable 24.2% occurrence rate when analyzing placenta and fetal tissues.
Investigations for uniparental disomy (UPD), notably involving chromosome 16, are advised due to their
association with a higher frequency of FGR and premature delivery. High mosaicism levels in chorionic villus
sampling (CVS) and UPD has been observed to result in adverse pregnancy outcomes (Eggenhuizen et al.,
2021)
Overall, in cases of suspected CPM, pregnancy should be classified as high-risk, warranting intensive fetal growth
monitoring from the first trimester, especially if trisomy involves chromosomes 2, 3, 7, 13, 15, 16, or 22. CPM especially
involving chromosomes 21 and 8 poses a heightened risk for FGR. Counseling future parents should involve emphasis on
the increased incidence of premature birth, structural fetal anomalies, FGR, and low birth weight. Conversely, CPM
involving trisomies for chromosomes 9, 10, 12, 18, and 20 does not show indications of adverse pregnancy outcomes
(Eggenhuizen et al., 2021)
Mosaicism in embryos detected by preimplantation genetic testing
With the advent of preimplantation genetic testing for aneuploidies (PGT-A), a new clinical scenario has emerged where
mosaicism is detected in embryos. Embryos with an intermediate result after PGT-A between the range of euploidy and
the range of aneuploidy, have been termed as ‘mosaic embryos’. An embryo that is putatively mosaic is a frequent finding
after PGT-A in recent times due to high sensitivity and resolution of NGS. A recent study showed a mosaic embryo rate
of 2-13% with NGS analysis in trophectoderm cells and tends to decrease throughout pregnancy (Popovic et al.,
2020)
Embryonic mosaicism can be classified using different parameters: grade of mosaicism (based on the percentage of
aneuploidy), number of chromosomes involved (simple mosaic, complex mosaic or chaotic mosaic), cell lines affected [total,
inner cell mass (ICM), trophoectoderm, ICM/trophoectoderm] or type of abnormality (whole-chromosome mosaic or
segmental mosaic).The clinical decisions around transferring this type of embryo can be challenging, particularly when no
chromosomally normal embryo is available.
Different international societies have published guidelines and position statements with their own set of
recommendations regarding transfer priority in cases of embryo mosaicism detected through preimplantation genetic
testing for aneuploidies (PGT-A). A summary of these recommendations is as follows:
a)
Mosaic euploid/ monosomy mosaicism should be prioritized over euploid/ trisomy mosaicism
[Preimplantation Genetic Diagnosis International Society (PGDIS) position statement 2016]
b)
Low mosaicism level (20-40%) should be prioritized over high-level mosaicism (40-70%) [Controversies
in Preconception, Preimplantation and Prenatal Genetic Diagnosis (CoGEN) 2017; PGDIS 2021 third
statement]
c)
Theoretical implications of the chromosome(s) involved should be considered (chromosomes not associated
with a known chromosomal disorder should be favoured over chromosomes associated with uniparental
disomy, intrauterine growth restriction or a viable chromosomal syndrome) (PGDIS 2016)
d)
Avoid the transfer of mosaic embryos involving chromosomes 13, 14, 16, 18, 21 or 45,X as they carried the
greatest risk of an affected viable pregnancy (PGDIS 2019 second statement, CoGEN 2017)
e)
Segmental mosaicism prioritized over whole-chromosome mosaicism (PGDIS 2021 third statement)
f)
When two embryos have identical qualities, a preference could be established based on their morphology
(PGDIS 2021 third statement)
g)
A new stimulation cycle is not advised when transferrable low-level mosaic embryos are available [European
Society of Human Reproduction and Embryology (ESHRE) 2022]
There are conflicting reports regarding the clinical outcome of transferring mosaic embryos. While some have
reported a reduced rate of implantation and an increased rate of miscarriage with mosaic embryo transfers,
others have reported successful pregnancies following the transfer of embryos with mosaicism. However,
data regarding the outcome for specific types of mosaicism are still limited and should be interpreted with
caution. Each mosaic embryo should be evaluated individually, and in cases involving more than one mosaic
embryo available for transfer, prioritization should be based on the most current evidence. According to
current evidence, embryos with results within the mosaic range should not be discarded or disregarded for
transfer, as this practice could negatively affect the cumulative live birth rate per cycle (Munoz et al.,
2024).
Overall, preimplantation genetic testing for aneuploidies (PGT-A) is not without its limitations, including the
potential for both false positive and false negative results. While some studies indicate a favorable impact of PGT-A on
pregnancy outcomes, particularly in specific cases, the results are inconsistent, especially for women under 35 years old.
Given these uncertainties, it is crucial to offer amniocentesis after the transfer of mosaic embryos to confirm the genetic
status of the fetus. Pretest counseling is a critical component of incorporating PGT-A into assisted reproduction. During
counseling, families should be thoroughly informed about the benefits, limitations, and potential risks associated with
PGT-A. They should also be made aware of their right to refuse PGT-A, ensuring that their decision is well-informed and
respects their autonomy.
Table 1: Genetic tests for detection of mosaicism (Campbell et al., 2015)
Technique
Clinical utility and Limitations
Karyotype
Cytogenetic analysis of 30 metaphase cells can identify 10% mosaicism with a confidence level of 95% in case of sex chromosomal mosaicism. Poor resolution (5-10Mb), Cannot detect mosaicism <10%.
FISH
Laborious; cannot detect mosaicism <10%; can count hundreds of cells in interphase FISH with relative ease.
Array-basedtechniques
aCGH: mosaicism detection level as low as 10¢20% SNP array: Mosaicism for CNVs of modest size can be detected as low as 5% by searching for slight deviation from expected allele frequencies, but resolution is limited by availability of known polymorphic SNPs.
Sangersequencing
Mosaicism for dominant alleles present in less than 25¢35% or greater than 65¢70% of cells can remain undetected.
Pyro-sequencing
Offers better minimum detection limits (as low as 5%) with more quantitative results. Results can be impeded by sequence features of the target, specifically repetition of the same nucleotide.
Massivelyparallelsequencing
The ability to detect mosaicism is proportional to the read depth of coverage or number of reads that are available covering a given base position. Next-generation sequencing with deep sequence coverage enhances sensitivity and allows for accurate quantification of the level of mosaicism. Although NGS technologies allow for low-grade mosaicism detection, number of samples to be tested per one patient is limited because of the high cost.
Personalisedassays
For massively parallel sequencing, custom-capture reagents, which enrich the DNA library to be sequenced for template molecules of interest allow for greater sensitivity. Digital PCR: Identify mosaic SNVs and indels at levels as low as 0.1%. Molecular inversion probes: Identify mutant DNA at levels as low at 0.5%.
Overview of challenges in genetic counselling in mosaicism
Mosaic phenotypes may have incomplete syndromic features, which may stay unnoticed, especially in a
low-grade mosaicism
Mutation load in the material tested does not necessarily correlate with the severity of disease.
Blood cells are an unstable source of genetic material given multiple rounds of self-renewal during
haematopoiesis. Multiple sources of primarily obtained DNA needs to be examined. Ectodermal tissues can
be sampled from buccal brushings or hair root bulbs, mesodermal tissues are available from blood or saliva,
while endodermal origin DNA is available from urothelial cells collected in urine samples. Levels of mosaicism
across tissues and body locations can show surprising variability, even within the same embryonic lineage. A
common example includes the i(12p) (Pallister Killian syndrome) which is not usually observed in cultured
peripheral blood Tcell lymphocytes analysed by Gbanding, but can generally be identified in cultured skin
fibroblasts in a mosaic state.
Low levels of somatic mosaicism may not be detectable by standard genetic sequencing and pure germline
mosaicism will not be detectable by testing of specimen types routinely available to diagnostic laboratories.
Usually more than one technique is needed to recognize mosaicism and an additional method, usually
different than the first one used, is required to confirm the result (Table 1).
Empiric recurrence risk estimates are only available for the most prevalent and well-studied diseases making
counselling challenging for other rare genetic disorders (Table 2).
Table 2: Overview of counselling regarding recurrence risk * (Adopted from Wallace et al., 2022)
Genetic disorder
Counselling
Chromosome disorders that frequently occur in mosaic form
Recurrence risk in sibs is expected to be low in the absence of a genetic predisposing factor (such as biallelic BUB1B pathogenic variants) in parents.
Mosaic disorders due to postzygotic de novo heterozygous variants
The risk of recurrence in subsequent offspring of the parents of an affected child is not increased compared to the general population.
Monogenic disorders that frequently occur in mosaic form
The risk that a parent with germline mosaicism will transmit the pathogenic variant to offspring is <50%. When transmitted, the variant will be constitutional, and the child will be severely affected.
Disorders associated with epigenetic mosaicism
The recurrence risk for siblings would be expected to be as in the general population.
References
1. Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013; 14:
307-320.
2. Brison N, et al. Predicting fetoplacental chromosomal mosaicism during non-invasive prenatal testing.
Prenat Diagn. 2018; 38: 258-266.
3. Brunner HG, et al. Brief report: reverse mutation in myotonic dystrophy. N Engl J Med. 1993; 328: 476-480.
4. Bushman DM, et al. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy
number in single neurons from sporadic Alzheimer's disease brains. Elife. 2015; 4: e05116.
5. CoGEN Position Statement on Chromosomal Mosaicism Detected in
Preimplantation Blastocyst Biopsies [Internet]. IVF-Worldwide. com; 2017
[cited 2024 May 22]. Available from: https://ivf-worldwide.com/cogen/oep/
publications/cogen-position-statement-on-chromosomalmosaicism-detected-in-preimplantationblastocyst-biopsies.html
6. Consoli C, et al. Gonosomal mosaicism for a nonsense mutation (R1947X) in the NF1 gene in segmental
neurofibromatosis type 1. J Invest Dermatol. 2005; 125: 463-466.
7. De Rycke et al. ESHRE survey results and good practice recommendations on managing chromosomal
mosaicism [Internet]. U.S. National Library of Medicine; [cited 2024 May 22]. Available from:
https://pubmed.ncbi.nlm.nih.gov/36349144/
8. Eggenhuizen GM, et al. Confined placental mosaicism and the association with pregnancy outcome and
fetal growth: a review of the literature. Hum Reprod Update. 2021; 27: 885-903.
10. Huisman SA, et al. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J Med Genet.
2013 ;50: 339-344.
11. Inaba T, Nagamachi A. Revertant somatic mosaicism as a cause of cancer. Cancer Sci. 2021; 112: 1383-1389.
12. Lai-Cheong JE, et al. Revertant mosaicism in skin: natural gene therapy. Trends Mol Med. 2011; 17:
140-148.
13. Le Fevre, et al. Atypical Angelman syndrome due to a mosaic imprinting defect: Case reports and review
of the literature. Am J Med Genet A. 2017; 173: 753-757.
14. Li M, et al. Trisomy 9 mosaic syndrome: Sixteen additional patients with new and/or less commonly
reported features, literature review, and suggested clinical guidelines. Am J Med Genet A. 2021; 185:
2374-2383.
15. Moog U, et al. Disorders Caused by Genetic Mosaicism. Dtsch Arztebl Int. 2020; 116: 119-125.
16. Mu±oz E, et al. Representing the Special Interest Group in Reproductive Genetics of the Spanish Society
of Fertility. To transfer or not to transfer: the dilemma of mosaic embryos - a narrative review. Reprod Biomed
Online. 2024; 48: 103664.
17. PGDIS, 2016. Preimplantation Genetic Diagnosis International Society (PGDIS) position statement on
chromosome mosaicism and preimplantation aneuploidy testing at the blastocyst stage. pgdis. org [Internet]
Available from: https://www.pgdis. org/docs/newsletter_071816.html.
18. Popovic M, et al. Chromosomal mosaicism in human blastocysts: the ultimate challenge of preimplantation
genetic testing? Hum Reprod. 2018; 33: 1342-1354.
19. Pyott SM, et al. Recurrence of perinatal lethal osteogenesis imperfecta in sibships: parsing the risk
between parental mosaicism for dominant mutations and autosomal recessive inheritance. Genet Med. 2011;
13: 125-130.
20. Raskind WH, et al. ADCY5-related dyskinesia: Comments on characteristic manifestations and
variant-associated severity. Mov Disord. 2017; 32: 305-306.
21. Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses.
Neurology. 2001; 56: 1433-1443.
23. Toutain J et al. Confined placental mosaicism revisited: Impact on pregnancy characteristics and outcome.
PLoS One. 2018; 13(4): e0195905.
24. Van Opstal D, et al. Origin and clinical relevance of chromosomal aberrations other than the common
trisomies detected by genome-wide NIPS: results of the TRIDENT study. Genet Med. 2018; 20: 480-485.
25. Wallace SE, et al. Resources for Genetics Professionals — Mosaicism. 2022 Oct 27. In: Adam MP, Feldman
J, Mirzaa GM, et al., editors. GeneReviews« [Internet]. Seattle (WA): University of Washington, Seattle;
1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK5854 55/Wallace SE, et al.